ウィルムス腫瘍(腎芽腫)は、小児に最もよくみられる腎悪性腫瘍であり、米国では毎年約500例が新たに診断されている。⽇本における発生頻度は低く、年間70~100例程度と推測されている。米国との人口比を考えると、日本では米国に比して発生率が少ない。
-
疫学
-
米国では、腎腫瘍の年間発生率は15歳未満の小児100万人当たり約7例で、小児悪性腫瘍全体の5%を占め、年間約500例が新たに発生する。ウィルムス腫瘍は15歳未満の小児に最もよくみられる腎悪性腫瘍で、全症例の約95%を占める。米国では、ウィルムス腫瘍の症例の3分の2は5歳未満、95%は10歳未満で診断される。対照的に、腎細胞がん(RCC)は15~19歳の年齢層に多い。
ほとんどの患者が孤立性のウィルムス腫瘍であり、5~7%が両側の腎臓に病変を有し、10%が単一の腎臓内に多巣性の病変を有する。片側性ウィルムス腫瘍の場合、診断時の年齢中央値は女児で43ヵ月、男児で37ヵ月である。両側性病変を有する小児は、より早い年齢で診断される(中央値は女児で31ヵ月、男児で24ヵ月)。無虹彩症や泌尿生殖器異常などの先天異常を伴う患者もまた、早期に診断される。
ウィルムス腫瘍の発症リスクは民族によって異なり、アフリカ系アメリカ人ではリスクが高く、アジア系ではリスクが低い。エピジェネティクスの違いは、アジア人の小児における発病率の低さに寄与している可能性があり、これはアジア人患者の腫瘍においてインスリン様成長因子2(IGF-2)のインプリンティング消失の頻度が低いことを報告した研究によって実証されている。
-
病因
-
ウィルムス腫瘍は、正常な尿細管および糸球体分化を伴わない後腎芽球の増殖をもたらす腎の発育異常によって引き起こされるようである。ウィルムス腫瘍は、ネフロジェニックレストまたはネフロブラストマトーシスと呼ばれる、持続性の後腎細胞の病巣から発生すると考えられている。neprogenic rest(異常に残留している胎生期の腎前駆細胞の細胞群)は通常、新生腎の1%にみられ、小児期には早期に退縮する。対照的に、片側性ウィルムス腫瘍の腎の35%、両側性疾患の腎のほぼ100%にみられる。
遺伝学的異質性
ウィルムス腫瘍は、小児期に最も一般的な腎臓がんであり、遺伝的な不均一性を持ちます。この病気には、複数の異なる遺伝子変異が関与していることが知られています。
XT1(194070):染色体11p13上のWilms tumor-1 (WT1)遺伝子のヘテロ接合性変異が原因。
WT2 (194071): 染色体11p15上のH19/IGF2インプリンティング制御領域(ICR1; 616186)の突然変異によって引き起こされます。
WT3 (194090): 染色体16qに位置する遺伝子座に関連しています。
WT4 (601363): 染色体17q12-q21にマップされた遺伝子座が関係しています。
WT5 (601583): 染色体7p14上のPOU6F2遺伝子(609062)の変異に起因します。
WT6 (616806): 染色体4q12上のREST遺伝子(600571)の変異が原因です。
また、ウィルムス腫瘍の発生には他の遺伝的要因も関わっています。例えば、BRCA2遺伝子(600185)の変異や、HACE1遺伝子(610876)のまれな体細胞および体質的破壊が報告されています。さらに、グリピカン-3遺伝子(GPC3;300037)の体細胞突然変異や、WTX遺伝子(300647)の体細胞突然変異もウィルムス腫瘍に関連しているとされており、特にWTX遺伝子の変異は男性では単一のX対立遺伝子上に、女性では活性型X対立遺伝子上に存在することが示されています。これらの様々な遺伝的要因がウィルムス腫瘍の発生に寄与しており、その遺伝的背景の多様性を反映しています。
遺伝学的病因
ウィルムス腫瘍は、多くの腫瘍抑制遺伝子および転写遺伝子の機能喪失変異と関連している。これらには、WT1、p53、FWT1、FWT2遺伝子および11p15.5遺伝子座の変異が含まれる。ウィルムス腫瘍の病因におけるこれらの遺伝子変異の役割は、まだ不明である。
- WT1遺伝子
- -WT1遺伝子は染色体11p13に位置する。WT1遺伝子産物は発育中の腎臓、精巣、卵巣で発現する。泌尿生殖器組織の発生と分化に関与しているようである。WT1遺伝子の変異は、ウィルムス腫瘍の小児で最初に同定された遺伝子異常であり、WAGR症候群の小児の核型分析で発見された。WAGR症候群の11p13欠失は、WT1およびPAX6(無虹彩症と関連)遺伝子を含むいくつかの連続した遺伝子を包含している。一方、Denys-Drash症候群の患者では、WT1遺伝子の8番目または9番目のエクソンに点変異があり、その結果、臨床所見が得られている。散発性ウィルムス腫瘍患者でWT1遺伝子変異を有するのは10%未満であり、他の機序が関与していることが示唆されている。
- 11p15.5-11p15.5遺伝子座
- 11p15.5-11p15.5遺伝子座は、WT2遺伝子座とも呼ばれ、インプリンティング遺伝子のクラスターが存在する。この遺伝子座の変異は、Beckwith-Wiedemann症候群(BWS)を含む、成長遅延または過成長を特徴とする多くの症候群で同定されている[12,18,31]。インプリンティング遺伝子とは、父方または母方に遺伝した遺伝子のコピーのどちらかが発現し、両方は発現しないような、親に由来する選択的遺伝子発現が、特定の組織で示される遺伝子のことである。例として、BWS患者では、Beckwith-Wiedemann遺伝子の母方コピーが配偶子形成時にサイレンシングされ、父方コピーのみが発現する。その結果、BWSの子孫は父親から受け継いだ突然変異を受け継ぎ、母親からベックウィズ・ヴィーデマン遺伝子の突然変異を受け継いだ人は無症候性保因者となり、突然変異を子孫に受け継ぐことができる。BWSと11p15遺伝子変異を持つ患者は、ウィルムス腫瘍のリスクが高い。
11p15の体細胞欠損は、ウィルムス腫瘍細胞、ウィルムス腫瘍に伴う腎周囲憩室、およびウィルムス腫瘍を取り囲む一部の正常な腎細胞で見つかっており、11p15の突然変異が非染色性のウィルムス腫瘍の発生において初期の役割を担っている可能性が示唆されている。さらに、ある研究では、非症候群性ウィルムス腫瘍患者の3%(437人中13人)およびウィルムス腫瘍を有する1家族のリンパ球において、この遺伝子座に由来する遺伝子の生殖細胞系列(体質)変異が証明され、220人の対照群では11p15の欠損は検出されなかった。体質性11p15欠損を有する患者は、11p15変異のない患者と比較して、両側腫瘍病変を有する可能性が高い。
- p53遺伝子
- TP53はヒト癌において最もよく変異する遺伝子であり、大腸癌、非小細胞肺癌、骨肉腫、ユーイング肉腫など様々な悪性腫瘍と関連している。p53遺伝子変異は、良好な組織型の腫瘍患者ではほとんどみられないが、未分化の組織型をもつウィルムス腫瘍の約75%にみられる。ある研究では、びまん性未分化癌におけるp53遺伝子変異の状態は、腫瘍の再発および死亡のリスク増加と関連していた。
- 家族性ウィルムス腫
- 家族性ウィルムス腫瘍は症例の1~2%を占める。遺伝様式は常染色体優性遺伝であるようで、浸透性は様々である。これらの家系では、WT1遺伝子変異との関連は認められない。17q12-21のFWT1遺伝子座、19q13.3-q13.4のFWT2遺伝子座、および11p15.5遺伝子座との連鎖が指摘されている。
-
病理学的特徴
-
ほとんどのウィルムス腫瘍は孤立性病変である。しかしながら、患者の5~7%が両側腎病変を有し、10%が1つの腎臓内に多巣性病変を有する。
ウィルムス腫瘍は、嚢胞、出血または壊死を含むことがある。腫瘍は一般的に仮性被膜に囲まれており、これが他の腎腫瘍(浸潤性の境界を有する)との鑑別に役立つことがある。
組織学的に、古典的な良好な組織型のウィルムス腫瘍は、3つの細胞型から構成されている。
芽球細胞-未分化細胞
間質細胞-未熟な紡錘形細胞および異種の骨格筋、軟骨、骨軟骨または脂肪。
上皮細胞-糸球体と尿細管ウィルムス腫瘍の中には、1種類または2種類の細胞しか含まないものもある。細胞型が1つしかない腫瘍では、ウィルムス腫瘍と診断することはしばしば困難である。
腫瘍の組織型は患者の転帰と関連している。未分化型は、多極性多角体有糸分裂像および高色素化を伴う顕著な核の拡大があることとして定義され、不良な転帰と関連している 。
-
臨床像
-
腎の胚性悪性腫瘍であるウィルムス腫瘍(腎芽細胞腫)は、小児期に最もよくみられる腎腫瘍である (Pizzo and Poplack’s Pediatric Oncology, 8 ed. Wolters Kluwer; 2021:673-92)。通常、一見健康な小児の腹部腫瘤として現れる。腹痛、発熱、貧血、血尿、高血圧は罹患児の25~30%にみられる。
ウィルムス腫瘍患者の約5%~10%が両側性または多中心性の腫瘍を有する。両側性腫瘍の有病率は、ウィルムス腫瘍の素因を有する個体では、遺伝的素因を有さない個体よりも高いが、片側性、単巣性のウィルムス腫瘍は、生殖細胞系列またはエピジェネティックな原因が根底にあることを排除しない。
ウィルムス腫瘍のほとんどの小児は、腹部の腫瘤または腫脹を呈するが、他の徴候や症状はみられない。その他の症状には、腹痛(患者の30~40%)、血尿(12~25%)、発熱、高血圧(25%)などがある。被膜下出血の患者の一部は、急激な腹部腫大、貧血、高血圧、時には発熱を呈することがある。肺は最も一般的な転移部位であるが、小児が呼吸器症状を呈することはまれである。
身体所見では、硬く圧痛のない滑らかな腫瘤が認められ、偏心した位置にあり、正中線を越えることはまれである。
ウィルムス腫瘍が疑われたら、強く触診すると腎被膜が破裂することがあるため、その後の腹部検査は慎重に行うべきである。。
ウィルムス腫瘍は先天異常および症候群を伴うことがあるため、検査には、無虹彩症、半側半球症、泌尿生殖器異常などの関連異常の評価を含めるべきである。
ウィルムス腫瘍の素因(罹りやすさ)を示唆する特徴
ウィルムス腫瘍の素因(罹りやすさ)を示唆する特徴としては、以下が挙げられる。
両側性ウィルムス腫瘍
片側の多中心性ウィルムス腫瘍
早期発症ウィルムス腫瘍(2歳未満)
多発性ネフロジェニックレスト(片側、両側とも)
ウィルムス腫瘍素因症候群の臨床的特徴:家族歴。ウィルムス腫瘍患者の約1%~2%は、少なくとも1人の親族がウィルムス腫瘍と診断されている。一部の家系では生殖細胞系列の病原性変異が同定されているが、約3分の2の個体ではまだ不明である。
-
サーベイランス
-
ウィルムス腫瘍患者は、ベックウィズ・ウィーデマン症候群BWSまたはWAGR症候群の小児などの高リスク患者のサーベイランスで見つけることができる。プログラムとしては、以下のように、連続腹部超音波検査によるサーベイランスが推奨される。
BWSまたは孤立性半数体形成不全の小児では、7歳まで3ヵ月ごと。
WAGR および WT1 関連症候群の小児では、5 歳まで 3 ヵ月ごと。
家族性ウィルムス腫瘍の同胞、両側ウィルムス腫瘍の生存者の子では、8歳まで3ヵ月ごと生殖細胞系列の病原性変異体またはウィルムス腫瘍関連症候群を有する小児のサーベイランス
一般的な考察 ウィルムス腫瘍の遺伝的素因を有する個体におけるサーベイランスの目標は、腫瘍の病期が低いうちに発見し、治療が少なくて済むようにすること、および/または進行期の腫瘍に比べて治療が成功しやすいようにすることである。経済的コスト、不必要な介入や心理社会的苦痛につながる偽陽性の可能性など、サーベイランスのリスクを考慮する。専門家は、ウィルムス腫瘍のリスクが1%以上~5%の人にサーベイランスを推奨している。しかし、推奨は地域によって異なるため、医療提供者は意思決定において地域の慣行を考慮すべきである。ウィルムス腫瘍は1週間ごとに大きさが倍増する可能性があるため、腹部超音波検査を3ヵ月ごとに実施すべきである。
ほとんどのガイドラインでは、ウィルムス腫瘍の90%~95%が発生する年齢までサーベイランスを継続することを推奨している。特定の症候群の患者においてウィルムス腫瘍のサーベイランスをいつまで継続すべきかを決定する1つの方法は、ウィルムス腫瘍の全リスクおよび年齢関連リスクを用いてウィルムス腫瘍の残存リスクを計算することである。例えば、WAGR症候群では、全ウィルムス腫瘍リスクは〜50%であり、腫瘍の90%は4歳までに発生し、腫瘍の98%は7歳までに発生するため、4歳の個体の残存ウィルムス腫瘍リスクは5%(0.5×0.1)であり、7歳の個体の残存ウィルムス腫瘍リスクは1%(0.5×0.02)である。従って、サーベイランスは、医療提供者、罹患者、および家族のリスク許容度に応じて、5~8歳まで継続される。
WT1およびBWSスペクトラム障害については、腫瘍サーベイランスの推奨について他のGeneReviewsの章を参照のこと。サーベイランスガイドラインが公表されているウィルムス腫瘍素因症候群(Li-Fraumeni症候群、DICER1症候群、体質性ミスマッチ修復欠損症 )の患者は、その症候群で推奨されているサーベイランスを受けるべきである。ウィルムス腫瘍のリスク年齢が確立されていないがん素因については、腹部超音波検査によるサーベイランスを8歳まで3ヵ月ごとに行うことが推奨される。
分子的原因が特定されていない両側性および/または多巣性のウィルムス腫瘍の患者
ウィルムス腫瘍に対する治療が完了した後は、8歳になるまで3ヵ月ごとに腎超音波検査による多発性腫瘍のスクリーニングを受けるべきである。最近の証拠によると、両側性のウィルムス腫瘍を有する個体は、ウィルムス腫瘍になりやすい生殖細胞系列の病原性変異体、または側性になる前の胚発生の初期に生じた体細胞病原性変異体のいずれかを有することが示唆されている。生殖細胞系列変異または早期体細胞変異のいずれかが、メタクロナスウィルムス腫瘍のリスクを増加させる。
ウィルムス腫瘍素因リスクのある親族に対するサーベイランス
複数のウィルムス腫瘍患者がいる家族、および両側性および/または多巣性のウィルムス腫瘍患者が1人いる家族では、迅速な治療開始および予防措置が有益となる親族をできるだけ早期に同定するために、リスクのある親族を評価することが適切である。
罹患家族において原因となる遺伝子変化が同定されている場合、評価には分子遺伝学的検査を含めることができる。原因遺伝子変異に関連するウィルムス腫瘍のリスクが十分に高い場合は、超音波検査によるサーベイランスが実施される。
罹患家族において原因となる遺伝子変化が同定されていない場合
罹患者の子孫は、8歳になるまで3ヵ月ごとに腹部超音波検査によるスクリーニングを受けるべきである。注意:両側性ウィルムス腫瘍の生存者の子供におけるウィルムス腫瘍のリスクは不明である。一部の両側性ウィルムス腫瘍は、体細胞またはエピジェネティックな変化から発生し、側性になる前の胚発生の初期に発生し、両方の腎臓に影響を及ぼすが、生殖細胞系列には存在しない。このような変異は遺伝しない。
罹患者の兄弟姉妹のサーベイランスの有益性は不明である。家族性ウィルムス腫瘍に関するNational Wilms Tumor Studyによると、両側ウィルムス腫瘍の456人中15人(3.3%)にウィルムス腫瘍の家族がおり、そのうち6人は3つの家族内に集まっていた。罹患している家族のほとんどは兄弟姉妹ではないため、家族性疾患が知られていない両側性ウィルムス腫瘍患者の兄弟姉妹にウィルムス腫瘍が発見される可能性は1%未満である。
-
診断・病勢評価
-
ウィルムス腫瘍の確定診断は、外科的切除または生検の際に組織学的に確認することによって行われる。
画像診断
画像診断は、ウィルムス腫瘍と他の腹部腫瘤の原因との鑑別に有用である。画像検査はまた、外科的アプローチや術前化学療法の必要性など、組織学的診断を確定する前の管理決定の指針にもなる。
腹部超音波検査は通常、腹部腫瘤の評価のために最初に行われる検査である。超音波検査は、腹部腫瘤または腫脹として現れる水腎症および多嚢胞性腎疾患を検出する。腎腫瘍が疑われる患者では、ドップラー超音波検査を行って腎静脈や下大静脈への腫瘍浸潤を検出し、血流の開存性を評価することができる。この情報はコンピュータ断層撮影(CT)でもチェックできる。
術前の破裂や腹水のチェックを含め、腫瘤の性質と範囲をさらに評価するには、造影CTまたは磁気共鳴画像法(MRI)が推奨される。CTまたはMRIはまた、超音波検査では検出されなかった反対側の腎臓の小さな腫瘍病変またはnepphrotic restを検出することもある。
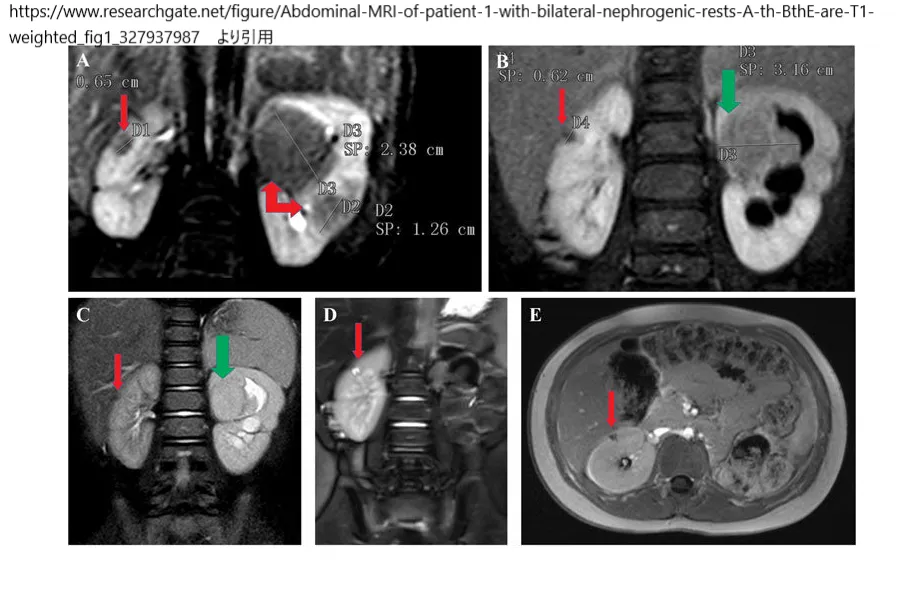
www.researchgate.net/figure/Abdominal-MRI-of-patient-1-with-bilateral-nephrogenic-rests-A-th-BthE-are-T1-weighted_fig1_327937987
より引用。両側腎性休息を有する患者1の腹部MRI。AþBþEはガドリニウム造影増強後のT1強調画像(AþBは冠状、Eは軸方向)、CþDは冠状T2強調画像(Cは脂肪飽和);(A)は両腎に腎性休息を示し(左腎に2つ、右腎に1つ)、2010年4月の初診時には左腎に最も大きなものがあった。(B þ C)2012年5月、左腎臓上極にウィルムス腫瘍(緑矢印)(水腎症)、右腎臓に小さな残存病変(赤矢印)。(D þ E)2016年3月、右腎(赤矢印)に小さな変化のない腎性残遺を示す。左:過去の腎摘出術。局所病期分類におけるCTとMRIの診断能においては、リンパ節転移および被膜貫通を検出する能力は同等であるため、小児腎腫瘍の初回局所病期分類にはいずれのモダリティも使用可能である。
術前画像診断は、診断時に腫瘍が手術可能かどうかを外科医が判断するのに役立つ。以下の所見または状況のいずれかを有する患者は、一般に、最初の一次腎摘除術ではなく、前もって生検を行い、腎摘除前の化学療法を受ける。
肝静脈以上の腫瘍血栓
巨大な腫瘍や広範な肺転移による肺機能低下
連続した構造物(副腎以外)の切除が必要な場合
腎摘除を試みると、重大な結果や腫瘍の流出、または腫瘍の残存を招くと外科医が判断した場合臨床検査
臨床検査には、尿検査を含む腎機能検査、肝機能検査、血清カルシウム検査、全血球算定検査、および凝固検査が含まれる。
後天性von Willebrand病は、診断時にウィルムス腫瘍患者の4~8%にみられるため、出血歴または多血性腫瘍の既往歴のある小児では、von Willebrand病用のvon Willebrand測定を含む凝固検査を受けるべきである。この異常は通常、臨床的意義はほとんどないが、過剰出血の既往歴のある患者は凝固異常症についてスクリーニングを受け、異常があれば周術期に情報共有すべきである。
病期分類
ウィルムス腫瘍の病期分類基準は、遺伝学的、組織学的、または生物学的マーカーを考慮することなく、腫瘍の解剖学的範囲に基づいている [79] 。病期が高いほど、より予後不良な広範囲な病変を示す。その結果、一般的に腫瘍の病期が高い患者には、より積極的な治療レジメンが投与される。(ウィルムス腫瘍の治療と予後」を参照のこと)。
現在使用されている主なステージングには2つある。外科的評価に対する化学療法のタイミングが異なるため、この2つのシステムを用いた試験の直接比較は困難である。
Children’s Oncology Group;COG
COG病期分類システムは、米国およびカナダ全域で使用されており、化学療法実施前の外科的評価に基づいている。最初の4つの病期は片側浸潤に限定される。
- I期:腫瘍は腎臓に限局している。腫瘍は無傷の被膜で完全に切除され、腎洞血管の浸潤や切除断端または切除断端を超える腫瘍は認められない。過去に破裂や生検がない。腫瘍がI期として特定の治療プロトコルに適格であるためには、所属リンパ節を顕微鏡で検査する必要がある。
- II期:腫瘍が腎臓を超えて進展しているが、切除断端または切除断端以遠に腫瘍を認めず、完全に切除されている。進展には、腎被膜を超える浸潤、腎洞軟部組織への浸潤、腎実質を超えるが切除標本内の血管浸潤が含まれる。腫瘍の生検を含む脇腹に限局した流出の破裂は、もはやII期には含まれず、現在ではIII期に含まれる。
- III期:手術後、残存腫瘍はあるが腹部内に限局している。これには、局所リンパ節転移、腹膜表面転移、腫瘍の不完全切除、切除断端または切除断端以遠の腫瘍、腫瘍の流出(spillage)、以前の生検、または腫瘍摘出前の術前化学療法が含まれる。
- ステージIV:血行性転移(肺、肝臓、骨、脳など)またはリンパ節転移が腹骨盤部を超えてある。
- V期:診断時に両側の腎病変を認める。診断時に両側の腎病変が存在する場合、その後の管理方針を決定するために、両側を別々に病期分類する。
国際小児腫瘍学会病期分類
SIOP病期分類はヨーロッパで広く用いられており、腫瘍を縮小する化学療法を行った後の外科的評価に基づいている。
- ステージ1:腫瘍は完全に切除され、腎臓に限局しているか、腎臓外にある場合は線維性仮性包に囲まれている。腫瘍は腎被膜、骨盤系(ただし尿管壁は含まない)、腎内血管に認められることがある。腎洞血管の浸潤や切除断端または切除断端以遠の腫瘍は認められない。
- ステージ2:腫瘍が腎臓または線維性仮性包を超えて進展しているが、切除断端または切除断端以遠に腫瘍を認めず、完全に切除されている。完全に切除された腫瘍浸潤は、腎実質、隣接臓器、大静脈を越えて腎洞、血管、リンパ節に進展することがある。
- ステージ3:手術後、残存腫瘍は残るが腹部に限局している。これには、局所リンパ節転移、腫瘍の不完全切除、腫瘍の腹膜表面への浸潤、切除断端の血管の腫瘍血栓、腫瘍の流出、または以前の生検が含まれる。血行性転移の証拠はない。
- ステージ4:血行性転移(例、肺、肝臓、骨、脳)またはリンパ節転移が腹骨盤を超えて認められる。
- ステージ5 – 診断時に両側腎病変を認める。
-
関連する先天性症候群
-
ウィルムス腫瘍は主に散発性疾患であり、ウィルムス腫瘍患者の1~2%のみが血縁者にウィルムス腫瘍患者がいる。約10%の症例において、ウィルムス腫瘍は、WAGR症候群、Denys-Drash症候群、Beckwith-Wiedemann症候群(BWS)などの多発奇形症候群の一部として発生する。
WAGR症候群
WAGR症候群は、ウィルムス腫瘍、無虹彩症、泌尿生殖器異常、および知的障害(精神遅滞)の症候群を指す。WAGR症候群の小児では、ウィルムス腫瘍を発症するリスクは約50%である。この症候群の小児は、11p13に位置するWT1遺伝子の遺伝子座の欠失を有する。WT1遺伝子の産物は生殖腺と腎臓の発達に関与する転写因子である。
WAGR症候群の小児54人のレトロスペクティブ研究では、以下の臨床所見が認められた。
無虹彩症:53人
泌尿生殖器の異常(例えば、陰睾、両性生殖器):41人
知的障害 :39人
ウィルムス腫瘍 :31人
腎障害(糸球体濾過量<80mL/minと定義)および蛋白尿:14人WAGR症候群患者64人を対象とした別のケースシリーズでは、全例が良好な組織像を有する腫瘍を有していた。11人(7%)が両側性であった。コホートの約半数に慢性腎疾患が発症し、その中には後に腎移植を受けた4人の患者の末期腎疾患も含まれていた。
デニス-ドラーシュ症候群 Denys-Drash syndrome
デニス-ドラーシュ症候群(ドラーシュ症候群)は、進行性腎疾患、男性仮性両性具有、ウィルムス腫瘍の三徴候である。罹患者はWT1遺伝子の8番目または9番目のエクソンに生殖細胞系列の点突然変異を有し、その結果、アミノ酸が置換され、ほとんど全ての患者(90%)がウィルムス腫瘍を発症する。
腎病理はびまん性メサンギウム硬化症で、乳児期に蛋白尿を呈し、ネフローゼ症候群、腎不全へと進行する。
ベックウィズ-ウィーデマン症候群 Beckwith-Wiedemann syndrome BWS
BWS患者は5~10%の確率でウィルムス腫瘍を発症する。BWSは、インプリンティング遺伝子群の部位である11p15.5領域のマイクロ重複変異によって引き起こされる。
BWSの主な臨床的特徴には、巨赤芽球症、巨舌症、小脳瘤、突出した目、耳のしわ、大きな腎臓、膵臓過形成、半身肥大などがある。
その他の先天異常
-その他の先天症候群や孤立性先天異常の患者もウィルムス腫瘍を発症するリスクがある。以下のようなものがある:
シンプソン-ゴラビ-ベーメル症候群
シンプソン-ゴラビ-ベーメル症候群は、グリピカン-3をコードする遺伝子の変異によって引き起こされるX連鎖性遺伝性疾患である。ウィルムス腫瘍を7.5%の確率で発症する。
ソトス症候群
ソトス症候群の小児では、ウィルムス腫瘍のリスクが2~3%ある。
パールマン症候群
パールマン症候群は、DIS3L2遺伝子の生殖細胞系列変異による常染色体劣性過成長症候群である。腎芽細胞腫を伴う両側腎過誤腫、ウィルムス腫瘍が特徴である。
家族性ウィルムス腫瘍
家族性ウィルムス腫瘍はまれで、BRCA2遺伝子またはTP53遺伝子の変異と関連している。
-
ウィルムス腫瘍の治療
-
治療には手術、化学療法、不完全切除や転移を有する患者に対する放射線療法がある。治療成績は一般に良好で、生存率は約90%と推定される。未分化型、両側性病変、遠隔転移、および再発病変は、あまり良好な転帰とはならない。
予後因子
初回診断時のいくつかの予後因子は、腫瘍の再発または死亡のリスク上昇と関連しており、以下のようなものがある。
腫瘍組織型
腫瘍の病期
分子マーカーおよび遺伝子マーカー: 染色体16q、1p、11p15におけるヘテロ接合性の消失(LOH)と1qの重複
年齢2歳以上これらの予後因子は、初期治療レジメンを決定する際に考慮される。
腫瘍組織型
腫瘍組織型は患者の予後と関連している。組織学的分類は、2つの主要なウィルムス腫瘍研究グループ、Children’s Oncology Group(COG)とInternational Society of Paediatric Oncology (SIOP)の間で多少異なっている。日本ではCOG方式で治療する施設が多い。
COGのプロトコールでは、化学療法前に組織学的評価が行われ、腫瘍は未分化かどうかによって分類される。
好ましい組織像(未分化型以外)
限局性未分化
びまん性未分化SIOPシステムでは、術前化学療法後の評価に基づいて組織学的分類を割り当てている。腫瘍は、腫瘍壊死の程度と3つの細胞型(上皮性、間質性、胚盤胞性)のそれぞれの相対的な割合に基づいて、低リスク、中間リスク、高リスクに分類される。化学療法後にびまん性未分化腫瘍または胚盤胞型腫瘍を認める患者は、高リスク組織型に分類される。
未分化型の存在は、ウィルムス腫瘍の小児における有害な転帰の最も重要な予測因子である。未分化型は、多極性多芽体有糸分裂像および高色素化を伴う著明な核拡大の存在として定義され、さらに限局性(原発腫瘍内の1ヵ所または数ヵ所に限局した未分化で、他の部位には未分化や著明な核異型を認めない腫瘍)またはびまん性に分類できる。未分化型ウィルムス腫瘍のほとんどがTP53遺伝子変異を有する。
National Wilms Tumor Study(NWTS)グループの報告では、診断時に非転移性疾患を有する患者632人の多変量解析により、未分化、ラブドイド、または明細胞肉腫の組織型が再発、転移、および死亡の割合の増加と関連していることが示された。びまん性未分化は患者の10%にしかみられなかったが、びまん性未分化腫瘍を有する患者は死亡例の60%以上を占めた。
治療方法
治療法は腫瘍の病期に基づいて以下のように決定される。
I期およびII期
外科的一次切除後、ビンクリスチンとダクチノマイシンを19週間投与する。放射線療法は必要ない。
以下のいずれかの患者では、治療法を変更できる。
超低リスク腫瘍-以下の基準をすべて満たす患者は、超低リスク腫瘍とみなされる。
年齢2歳未満
I期の良好な組織型
腫瘍の重さが550g未満である。超低リスク腫瘍患者のほとんどは、補助化学療法を行わずに腎摘除術のみで管理できる。
高リスク分子マーカー
上述したように、1p、11p15、16q染色体におけるLOHと1q重複などある種の分子マーカーは、それ以外の組織型が良好な患者における OS と EFS の低下と関連している(上記の’分子マーカー’を参照)。予備的なデータでは、これらの高リスクマーカーを持つ患者に対して治療を強化することで、予後が改善する可能性が示唆されている。例えば、1p染色体と16q染色体にLOHを合併した標準リスクのI期およびII期の腫瘍患者35人を対象とした単群試験では、化学療法レジメンにドキソルビシンを追加することは、ドキソルビシンを追加せずに治療を受けた以前の試験に登録された同様の患者と比較して、4年EFSが改善する有意でない傾向と関連していた。増強療法が1q増多を示す腫瘍患者の予後を改善するかどうかは不明である。この疑問を解決するにはさらなる研究が必要である。
未分化組織
I/II期の未分化組織の患者は予後不良であり、化学療法と放射線療法が追加される。
III期
外科的一次切除後、ビンクリスチン、ダクチノマイシン、ドキソルビシンの3剤併用化学療法を25週間行う。リンパ節転移または腹膜汚染の程度に応じた放射線療法(10.8Gy)を、罹患した脇腹、半腹部または全腹部に対して行う。III期の良好な組織像を有する患者のほとんどは、この治療法で良好な転帰を示す。リンパ節転移陽性と1p染色体または16q染色体のLOHは、予後不良の予測因子である。
1p染色体と16q染色体のLOHが合併している患者は、シクロホスファミドとエトポシドを含むより強力な化学療法レジメンMで治療される。
IV期
一次外科切除後、ビンクリスチン、ダクチノマイシン、ドキソルビシンの3剤併用化学療法を25週間行う。III期と同様に、染色体1pと16qのLOHが合併している患者には、レジメンMが行われる。放射線療法は、6週間の化学療法後も肺転移が完全に消失しない場合にのみ、肺野全体に行われる。
肺門および/または傍大動脈リンパ節への病巣転移が確認された患者には、適応に応じて腹部の適切な2分の1を含む局所領域に放射線療法(線量10.8Gy)を行う。肺以外に転移がある場合は、部位によって照射量が異なる。
ステージV
SIOPプロトコールは、患者は4週間の術前化学療法(転移が認められる場合は6週間)の後、外科的切除を行い、初回化学療法後に実施される病期分類および組織学的評価に基づく術後化学療法を行う。
非転移性疾患の患者に対しては、術前化学療法はビンクリスチンとダクチノマイシンを4週間投与する。腹膜-腹膜領域を超える転移性病変(例えば、肺転移)の証拠がある場合は、3剤併用化学療法(ビンクリスチン、ダクチノマイシン、ドキソルビシン)を術前に6週間行う。術前化学療法後、外科的切除と病期分類が行われる。
術後
術後のSIOPレジメンは、腫瘍の病期分類と組織型に基づいて以下のように設定されている。
- ステージI:
- -低リスク-低リスクのI期腫瘍(すなわち、初回術前化学療法後に残存腫瘍がない)の患者は、追加の化学療法または放射線療法を必要としない。これらの患者は再発について注意深く監視される。
-中等度リスク-治療は術後化学療法(ビンクリスチンおよびダクチノマイシン)を4週間行い、放射線療法は行わない。
-高リスク – 術後化学療法(ビンクリスチン、ダクチノマイシン、ドキソルビシン)を27週間実施し、放射線療法は行わない。 - ステージII:
- -低リスクおよび中リスク – 術後化学療法(ビンクリスチン、ダクチノマイシン)を27週間行い、放射線療法は行わない。以前はSIOPの治療プロトコールに含まれていたが、術後レジメンからドキソルビシンを除いても、中等度リスク組織型の患者のEFSを有意に悪化させることはなく、5年OSにも影響はなかった。
-高リスク-治療は、34週間の術後化学療法(ドキソルビシン、シクロホスファミド、カルボプラチン、エトポシド)と放射線療法(リンパ節転移または肉眼的病変に対して10.8Gyのブーストを伴う25.2Gyの脇腹放射線療法)である。 - ステージIII:
- -低リスク:術後化学療法(ビンクリスチン、ダクチノマイシン)を27週間実施し、放射線療法は行わない。
-中等度リスク:術後化学療法(ビンクリスチンおよびダクチノマイシン)を27週間実施し、放射線療法(リンパ節転移または肉眼的病変に対して10.8Gyのブーストを伴う14.4Gyの脇腹放射線療法)を行う。II期の腫瘍と同様に、術後レジメンからドキソルビシンを除いても、中等度リスクの組織像を有する患者の転帰を有意に悪化させることはなかった。-高リスク-II期の高リスク腫瘍と同様に、治療は34週間の術後化学療法(ドキソルビシン、シクロホスファミド、カルボプラチン、エトポシド)と放射線療法(リンパ節転移または肉眼的病変に対して10.8Gyのブーストを伴う25.2Gyの脇腹放射線療法)からなる。
- ステージIV
- 最初の6週間の術前化学療法後の肺結節の組織型と反応性に基づいて治療が行われる。
-低リスクおよび中リスクの組織型で完全奏効(すなわち、肺結節の消失または切除)-治療は術後化学療法(ビンクリスチン、ダクチノマイシン、ドキソルビシン)を27週間行い、肺への放射線照射は行わない。局所腫瘍III期の患者には脇腹放射線療法を行う。
-不完全奏効を示す低リスクおよび中リスク組織型-治療は、34週間の術後化学療法(ドキソルビシン、シクロホスファミド、カルボプラチン、エトポシド)と放射線療法(局所III期腫瘍に対しては15Gyの肺放射線療法と脇腹放射線療法)からなる。
-高リスク組織型-治療は、34週間の術後化学療法(ドキソルビシン、シクロホスファミド、カルボプラチン、エトポシド)と放射線療法(III期の局所腫瘍に対しては15Gyの肺放射線療法と脇腹放射線療法)である。 - ステージV
- 両側(ステージV)のウィルムス腫瘍の治療について以下に概説する。
特殊な集団-両側(V期)のウィルムス腫瘍、退形成性組織型、および/または肺転移を有する患者は一般に、標準リスクの組織型を有する低悪性度腫瘍の患者よりも集中的な治療を必要とする。両側腎病変-両側ウィルムス腫瘍(V期)の患者(表2)には、他のレジメンではなく、ビンクリスチン、ダクチノマイシン、およびドキソルビシンによる術前化学療法を推奨する。術後療法では、より広範な両側腎切除よりも腎実質温存切除を勧める。術後化学療法は、ネオアジュバント化学療法に対する治療効果、病理組織像、腫瘍の病期に基づいて、放射線を併用するかしないかを選択する。術前の併用化学療法は、早期のネフロン温存手術を容易にし、腎実質を温存することにより、末期腎不全のリスクを減少させる。
COGプロトコールでは、術前化学療法はビンクリスチン、ダクチノマイシン、およびドキソルビシンの6週間レジメンからなる。術後化学療法は、術前化学療法に対する臨床的および組織学的反応、病理学的分類、および腫瘍の病期を統合したリスク適応プロトコールに基づいて、放射線を併用する、または併用しない方法で行われる。
ウィルムス腫瘍患者の5~7%がV期(両側腎臓病変)を呈するが、その治療は困難である。このような患者の治療目標は、腎機能を温存しながら両側の腫瘍部位を適切に治療することである。
集中的化学療法を行った後に腎実質を温存する切除戦略をとることで、転帰が改善することが示された。COGが実施したプロスペクティブ研究(AREN0534)では、両側ウィルムス腫瘍の評価可能な患者189人に6~12週間の術前化学療法を行い、その後腫瘍切除を行った。この研究では、大半の患者(84%)が化学療法開始後12週間までに確定的な外科的治療を受けた。ほとんどの患者は、対側腎部分切除を伴う片側腎全摘術(48%)または両側腎部分切除術(35%)を受けた。対照的に、両側腎全摘除術を施行した患者は3%未満であった。4年EFSとOSは、それぞれ82%と95%であり、過去の対照群(8年EFSは40~74%、8年OSは45~89%)よりもかなり高かった。レトロスペクティブ研究では、ネオアジュバント化学療法についても同様の結果が報告されており、腎不全の発生率が減少し、患者の全生存率は最初の外科的切除を受けた患者と同等であった。
びまん性無形成(高リスク腫瘍)の患者は、術前化学療法を使用しても予後不良である。COG AREN0534のその後の解析では、180人の評価可能な患者が、術前化学療法に対する病理組織学的反応に基づいてリスク分類された。この研究において、4年EFSおよびOSはそれぞれ、低リスク症例および完全に壊死した腫瘍の症例(それぞれ100%)、中リスク症例(82%および97%)、および高リスクのblastemal型腫瘍の症例(79%および93%)で最も高かった。びまん性未分化の患者の生存率は最も低かった(4年EFSとOSはそれぞれ61%と72%)。同様の結果が、両側性腫瘍の小児80人を対象とした別の研究でもみられ、びまん性未形成または胚盤胞型腫瘍の患者では、集中的な術前化学療法はOSの改善と関連していなかった。
未分化型:
未分化型の患者は転帰が不良である。その結果、退形成性ウィルムス腫瘍患者において、代替薬(例えば、イリノテカン)を検討し、より集中的な治療を行う試験が実施されているが、4年EFSは不良である。びまん性未分化ウィルムス腫瘍患者のEFSを改善する、より効果的なレジメンを見つけることを期待して、現在進行中の臨床試験があり、将来の試験も計画されている。現在進行中のCOG研究(AREN1921)では、II期からIV期のびまん性未分化ウィルムス腫瘍に対する「レジメンUH-3」が研究されている。UH-3レジメンは、ビンクリスチン/ドキソルビシン/シクロホスファミド、シクロホスファミド/カルボプラチン/エトポシド、およびビンクリスチン/イリノテカンを交互に投与するサイクルで構成され、合計40週間持続する。
肺転移:
ウィルムス腫瘍患者の胸部画像診断は論争の的である。造影CTは胸部X線撮影よりも感度が高い。しかし、CTで検出された結節のうちどれだけが悪性疾患であり、これらの病変を診断することが転帰を改善するかどうかは不明である。肺転移を有する患者では、最適な管理は依然として不明確である。
COGの研究では、組織型が良好で孤立性肺転移を有する患者で、6週間の化学療法(ビンクリスチン、ダクチノマイシン、およびドキソルビシン)後に肺転移の完全な消失が認められた場合、肺照射を行わずに同じ化学療法レジメンを継続した。6週間の化学療法で効果が不十分であったか、または1pおよび16q染色体のLOHが認められた患者には、全肺放射線療法とシクロホスファミドおよびエトポシドの追加療法(レジメンM)が行われた。これらの結果は、初回化学療法に対する患者の反応性に合わせて治療を調整する戦略が、治療に関連した罹患率を減少させながら、肺転移を有する小児の優れた転帰を維持できることを示唆している。
SIOP 93-01試験では、肺切除前化学療法(ビンクリスチン、ダクチノマイシン、ドキソルビシン/エピルビシン)を6週間施行しても効果が不十分な患者は、可能であれば肺結節の外科的切除を受けた。6週間の術前化学療法後に肺転移が完全に消失した患者および肺結節の外科的切除が成功した患者には、その後、肺照射を行わずに術後化学療法(ビンクリスチン、ダクチノマイシン、およびドキソルビシン/エピルビシンを27週間)を行った。不完全切除の肺結節、多発性の手術不能転移、および/または原発腫瘍の高リスク組織像を有する患者には、より強力な腎摘除後化学療法レジメン(ドキソルビシン/エピルビシン、シクロホスファミド、カルボプラチン、およびエトポシドを34週間)を行った。9週間後に完全奏効が得られた場合、肺照射は行われなかった。残存肺結節および/または高リスク組織像を有する小児では、両側肺放射線療法が行われた。本試験では、初回治療で肺放射線照射を必要とした患者はわずか14%であった。5年EFSは73%、OSは82%であった。5年OSは、6週間の腎摘除前化学療法後に完全奏効を示した患者と転移切除を受けた患者で、不完全奏効を示した患者と比較して優れていた。
腫瘍再発に対する救済療法
再発は、組織型が良好な患者の約15%にみられる。未分化型の特徴を有する患者では、再発のリスクはかなり高い。ウィルムス腫瘍患者に対する最適な救済療法レジメンはまだ確立されていない。シクロホスファミド、イホスファミド、カルボプラチン、エトポシド、およびシスプラチンを用いた多剤併用レジメンが、程度の差こそあれ使用されている。
幹細胞レスキューを伴う大量化学療法は、再発腫瘍患者、特に予後不良因子を有する患者に対する選択肢として示唆されている。しかしながら、このアプローチの有効性は依然として不明である。

この記事の筆者:仲田洋美(医師)






