疾患に関係する遺伝子/染色体領域
疾患概要
Hypoaldosteronism, congenital, due to CMO II deficiency CMO II欠損による先天性低アルドステロン症 610600 AR 3
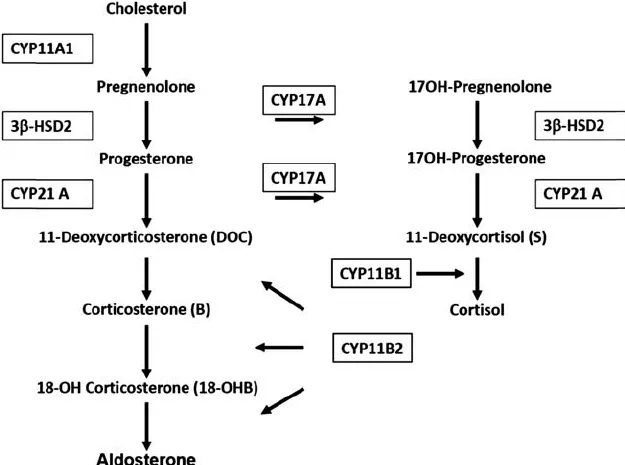
コルチコステロンメチルオキシダーゼII型(Corticosterone Methyl Oxidase Type II、略してCMO II)は、副腎皮質の糸球体層にある特定の酵素で、アルドステロンの生合成における最終段階を担います。具体的には、18-ヒドロキシコルチコステロン(18-OHB)からアルドステロンへの変換を触媒します。この酵素は、アルドステロン生合成経路において非常に重要な役割を果たし、体内のナトリウムとカリウムのバランス調節、それに伴う血圧の維持に必須です。
CMO IIの活性は、アルドステロンの産生を最終的に可能にするために、前駆体である18-OHBを最終的なアルドステロンへ変換することにより、体内の電解質バランスと血圧調節に直接的な影響を与えます。レニン-アンギオテンシン系や血中のカリウム濃度の変化など、複数の刺激に応じて調節されます。
CMO II型欠損症(CMO II Deficiency)は、CYP11B2遺伝子の変異により発症する遺伝性疾患であり、この酵素の機能不全が原因でアルドステロンの合成が不十分になります。CMO II型欠損症の患者では、18-OHBの過剰蓄積とアルドステロンの不足が見られ、低アルドステロン症や高レニン血症、塩分喪失による低血圧などの症状が発生します。これらの症状は、ナトリウムの保持不足とカリウムの過剰排泄によるものです。
この疾患は、特に新生児期や幼児期に重要な塩分バランスの維持に関わるため、早期診断と適切な治療が必要です。治療には、アルドステロンの不足を補うためにミネラルコルチコイドの補充や食塩摂取の増加が含まれます。CMO II型欠損症の理解と管理は、副腎皮質機能障害を持つ患者の生命を守り、生活の質を改善するために重要です。
コルチコステロン・メチルオキシダーゼII型欠損症(CMO II欠損症)は、CYP11B2遺伝子の変異によって発生する、常染色体劣性遺伝性疾患です。この病気は、アルドステロン生合成の生化学的最終段階である18-ヒドロキシコルチコステロン(18-OHB)からアルドステロンへの18-水酸化反応が欠如していることにより特徴づけられます。この反応の不在は、アルドステロンの産生不足と、結果として生じる塩分の損失を引き起こします。CMO II欠損症の患者では、アルドステロンのレベルは低いか正常ですが、18-OHBの分泌が増加し、コルチコステロンと18-OHBの比率が低下します。
一方、CYP11B2遺伝子産物は、アルドステロン生合成の初期段階においても機能し、コルチコステロンから18-OHBへの18-水酸化反応を触媒します。この酵素活性の不足は、CMO I型欠損症(別の常染色体劣性遺伝性疾患)を引き起こしますが、CMO I型とCMO II型は、生化学的特徴が異なるものの、アルドステロン合成経路における異なる段階に影響を与える点で表現型が重複します。CMO I型欠損症では、アルドステロンが検出されない場合が多いですが、18-OHBは低いか正常レベルを示します。
これらの疾患は、アルドステロン生合成経路におけるCYP11B2遺伝子の重要性を示しており、特定の遺伝子変異がどのようにして特定の代謝経路の障害につながるかを理解する上で有用です。適切な診断と治療を行うことで、CMO II欠損症およびCMO I欠損症の患者の塩分バランスの維持と生活の質の改善が可能になります。
臨床的特徴
Davidら(1968)は、アルドステロン欠乏症による成長遅延を伴うプエルトリコ人の兄妹を報告しました。症状は軽微で、一過性の血清電解質異常が見られました。この欠損は18-ヒドロキシコルチコステロンからアルドステロンへの最終変換に関わっていました。
Rappaportら(1968)は、アルドステロン減少による塩類喪失症候群を持つ2人の兄弟を観察し、18-OH-デヒドロゲナーゼの欠損を仮定しました。
Roslerら(1973,1977)は、イラン系ユダヤ人家族の小児における重度の食塩喪失症候群を報告しました。これはアルドステロン生合成経路の末端部分の先天性エラーによるもので、18-ヒドロキシコルチコステロンの顕著な過剰産生が特徴でした。
Ulick(1976)は、2型コルチコステロン・メチルオキシダーゼ欠損症(CMO II型)を提案しました。これは新生児期の高カリウム血症、低ナトリウム血症、代謝性アシドーシス、成長遅延が特徴です。
Veldhuisら(1980)は、CMO II型欠損症の北米人家族を報告しました。この欠損は食塩の補充で改善しました。
Leeら(1986)は、CMO II型欠損症の兄弟を調査し、ミネラルコルチコイド補充により正常値まで低下した18-OHBとアルドステロンの比を報告しました。
Hauffaら(1991)は、生命を脅かす塩分喪失と高カリウム血症を呈したCMO II型の2人の男児を報告しました。
Piccoら(1992)は、CMO II欠損症による反復性脱水と重度の発育不全の症例を報告しました。9-α-フルオロヒドロキシコルチゾンによる治療は成功しました。
これらの研究は、アルドステロン生合成の異常が引き起こす臨床的特徴と、遺伝的背景に関する重要な情報を提供しています。特にCMO II型欠損症は、新生児期から幼児期にかけての塩分喪失、高カリウム血症、成長の遅延が特徴であり、適切な治療により改善可能であることが示されています。
マッピング
一方、Mayerovaら(1991年)はドイツ人家族におけるCMO II型欠損症の症例でMspI制限酵素分析を用い、CYP11B1遺伝子の突然変異を除外しました。これは、CYP11B1遺伝子に関連する変異がCMO II型欠損症の原因とは限らないことを示唆しています。
これらの研究は、遺伝子マッピングと分子遺伝学的分析が遺伝性疾患の理解においていかに重要であるかを示しています。特に、同じ遺伝子または遺伝子近傍の異なる変異が異なる疾患の原因となり得ること、そして特定の遺伝子変異が特定の人口集団において共通している可能性があることが明らかになりました。このような知見は、遺伝性疾患の診断、予防、治療法の開発において貴重な情報を提供します。
分子遺伝学
Peterらによる別の研究では、Hauffaらにより報告されたCMO II型欠損症の患者から、CYP11B2遺伝子の別のホモ接合体変異(124080.0007)が同定されました。これは、CYP11B2遺伝子の変異がCMO II欠損症の原因であることを裏付けるものです。
CMO II欠損症は、アルドステロンの生合成経路における特定のステップに影響を与える遺伝的障害であり、CYP11B2遺伝子はこの過程で重要な役割を担っています。これらの研究により、特定の遺伝子変異がどのようにして特定の代謝異常を引き起こすかについての理解が深まり、遺伝性疾患の診断や治療に向けた重要な情報が提供されました。
疾患の別名
ALDOSTERONE DEFICIENCY II
HYPERRENINEMIC HYPOALDOSTERONISM, FAMILIAL, 1; FHHA1B
ALDOSTERONE DEFICIENCY DUE TO DEFICIENCY OF STEROID 18-OXIDASE
STEROID 18-OXIDASE DEFICIENCY
18-OXIDASE DEFICIENCY
CMO II欠乏症
アルドステロン欠乏症II
高レニン血性低アルドステロン症、家族性、1
ステロイド18-オキシダーゼ欠損によるアルドステロン欠乏症
ステロイド18-オキシダーゼ欠損症
18-オキシダーゼ欠損症







