疾患に関係する遺伝子/染色体領域
疾患概要
Hypoaldosteronism, congenital, due to CMO I deficiency CMO I欠損による先天性低アルドステロン症 203400 AR 3
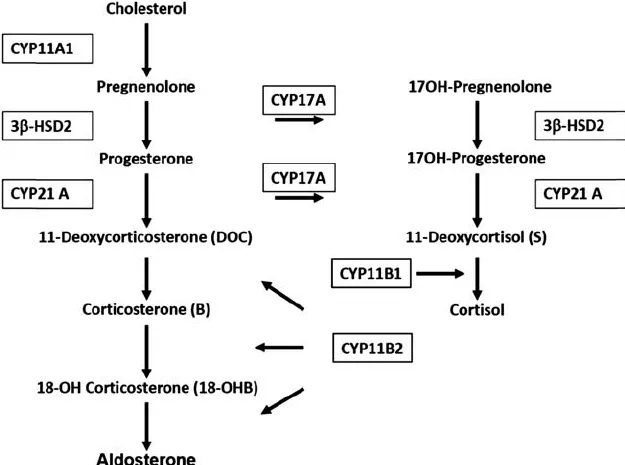
コルチコステロンメチルオキシダーゼI(Corticosterone Methyl Oxidase Type I、略してCMO I)は、副腎皮質の糸球体層に位置する特定の酵素です。この酵素は、アルドステロンの合成経路において重要な役割を果たし、具体的にはコルチコステロンから18-ヒドロキシコルチコステロンへの変換を担います。アルドステロンは、ナトリウムの再吸収とカリウムの排泄を促進することにより、血圧の調節と体液の電解質バランスの維持に不可欠なミネラルコルチコイドホルモンです。
CMO Iは、アルドステロン合成の複数段階のプロセスの一部であり、この段階ではコルチコステロンが酵素の作用によって酸化され、18-ヒドロキシコルチコステロンという中間体が生成されます。その後、さらなる変換過程を経て、最終的にアルドステロンが合成されます。
この酵素の活性は、レニン-アンギオテンシン系の刺激に応じて調節されることが多く、血中のナトリウム濃度が低下したり、カリウム濃度が上昇したり、血圧が低下すると活性化されます。これにより、アルドステロンの産生が促進され、ナトリウムの再吸収とカリウムの排泄が進み、血圧が上昇します。
CMO Iは、その特異的な機能と重要性から、ホルモン合成経路の理解、特に高血圧症や副腎機能障害などの疾患の研究において注目されています。また、CMO Iの活性異常は、アルドステロン合成異常症などの特定の病態の原因となることがあります。
コルチコステロン・メチルオキシダーゼI型欠損症(CMO I欠損症)は、CYP11B2遺伝子の変異によって引き起こされる常染色体劣性遺伝病です。この病気は、アルドステロンの生合成過程の最終段階である、コルチコステロンから18-ヒドロキシコルチコステロン(18-OHB)への18-ヒドロキシル化反応が行われないことが特徴です。この反応の欠損により、アルドステロンの産生が減少し、結果として塩分消耗の症状が現れます。CMO I欠損症の患者では、アルドステロンが検出されず、その直接の前駆体である18-OHBのレベルは低いか正常であり、コルチコステロンと18-OHBの比率が異常に高くなります。
一方、CYP11B2遺伝子産物は、18-OHBからアルドステロンへの変換を触媒する酵素でもあります。この酵素反応の欠損は、CMO II型欠損症(610600)という別の常染色体劣性遺伝病を引き起こします。CMO II型欠損症はCMO I型と表現型が重複する部分がありますが、生化学的特徴は異なります。CMO II型では、アルドステロンのレベルは低いか正常ですが、18-OHBの分泌が代償的に増加します。その結果、これらの患者ではコルチコステロンと18-OHBの比率が低くなります。
これらの病態は、アルドステロンの生合成におけるCYP11B2遺伝子の重要性を浮き彫りにし、特定の遺伝子変異がいかにして特定のホルモンの合成異常に繋がるかを示しています。これらの疾患の診断と治療には、遺伝子レベルでの理解が不可欠です。
臨床的特徴
Peterら(1997年)の追跡調査では、家族メンバーの血漿中アルドステロンと18-OH-コルチコステロンが減少し、血漿中コルチコステロンと11-デオキシコルチコステロンが増加していることが確認されました。これらの所見は、コルチコステロンの18-水酸化の欠損を示しており、CMO I型欠損症の診断につながりました。
Dropら(1982年)は6例のCMO I型欠損症の症例を報告しています。
また、Kayes-Wandoverら(2001年)は、47歳の男性がバリウム浣腸の準備中に高カリウム血症を発症し、CMO I型欠損症と診断された症例を報告しました。この男性は、乳児期の発育不全が特に顕著であったとされます。臨床検査では血清レニンの増加、アルドステロンの血清および尿中濃度の低下、尿中コルチコステロンの増加、および尿中18-ヒドロキシコルチコステロンの減少が確認されました。
分子遺伝学
さらに、VisserとCost(1964)によって報告されたCMO I型欠損症の患者2人において、Peterら(1997)はCYP11B2遺伝子にホモ接合性の変異を同定しました。この場合、罹患していない両親4人全員がその変異のヘテロ接合体キャリアでした。これは、この疾患がオートソーマル劣性遺伝であることを示しています。
また、Kayes-Wandoverら(2001)は、中年期にCMO I型欠損症の症状を示した男性患者において、CYP11B2遺伝子にホモ接合性の6ベースペア重複を同定しました。これらの研究は、CYP11B2遺伝子の異なる変異がCMO I型欠損症の原因となることを示しており、遺伝子診断や治療法の開発に貢献する重要な情報を提供しています。
CMO I型欠損症は、アルドステロン合成の最終段階を担う酵素の異常により発生します。アルドステロンは血圧と体内の電解質バランスの調節に重要な役割を果たすホルモンです。この疾患の患者は、塩分を保持する能力が低下し、低血圧や電解質バランスの乱れを示すことがあります。
疾患の別名
ALDOSTERONE DEFICIENCY I
HYPERRENINEMIC HYPOALDOSTERONISM, FAMILIAL, 1; FHHA1A
ALDOSTERONE DEFICIENCY DUE TO DEFECT IN STEROID 18-HYDROXYLASE
18-HYDROXYLASE DEFICIENCY
STEROID 18-HYDROXYLASE DEFICIENCY
CMO欠乏症
アルドステロン欠乏症I
高レニン血性低アルドステロン症、家族性、1;fhha1a
ステロイド18-ヒドロキシラーゼ欠損によるアルドステロン欠乏症
18-ヒドロキシラーゼ欠損症
ステロイド18-ヒドロキシラーゼ欠損症







