承認済シンボル:DYSF
遺伝子名:dysferlin
参照:
HGNC: 3097
AllianceGenome : HGNC : 3097
NCBI:8291
Ensembl :ENSG00000135636
UCSC : DYSF (ENST00000258104.8) from GENCODE V47
遺伝子OMIM番号603009
●遺伝子のlocus type :タンパク質をコードする
●遺伝子のグループ:Ferlin family
●遺伝子座: 2p13.2
●ゲノム座標: 2:71,453,561-71,686,763
遺伝子の別名
dysferlin, limb girdle muscular dystrophy 2B (autosomal recessive)
dystrophy-associated fer-1-like 1
fer-1-like protein 1
FER1L1
FLJ00175
FLJ90168
LGMD2B
遺伝子の概要
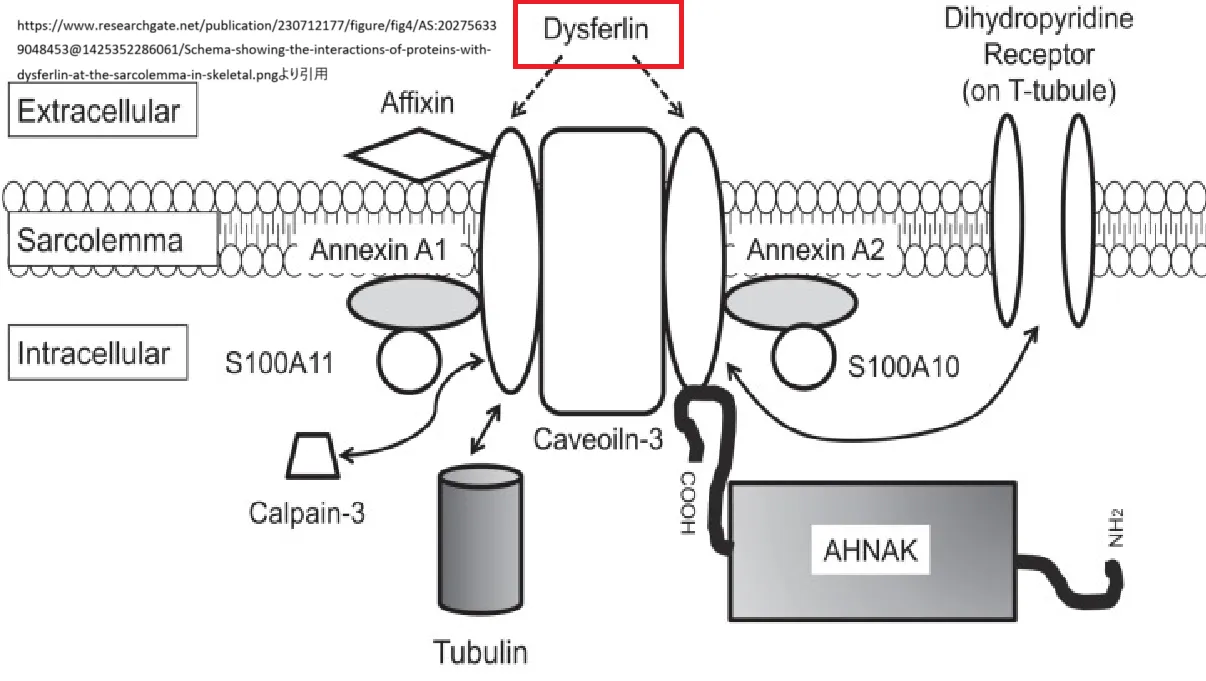
● ジスフェルリンの主な機能
1. 筋膜の修復:
筋肉が緊張や運動により損傷すると、筋膜に小さな裂け目や損傷が生じます。ジスフェルリンはこの損傷を感知し、カルシウム依存的な機構を通じて膜を修復します。この修復機能により、筋細胞が破壊されず、正常に機能し続けることができます。
2. 筋再生の可能性:
研究では、ジスフェルリンが新しい筋繊維の形成(再生)にも関与している可能性が示されています。これは、筋繊維が損傷した際に、新しい筋細胞が作られ、修復されるプロセスです。ジスフェルリンの欠損がこの再生プロセスにどのような影響を与えるかについては、まだ完全には解明されていません。
3. 炎症への関与:
ジスフェルリンは、炎症反応にも関与していると考えられています。筋膜の損傷後には、免疫系が修復プロセスに関与し、炎症反応を引き起こしますが、ジスフェルリンがこのプロセスの調節に何らかの役割を果たしている可能性があります。しかし、この機能に関しても、まだ十分に理解されていない部分があります。
● DYSF遺伝子の変異と疾患
DYSF遺伝子の変異は、肢帯型筋ジストロフィー2B型やミオパチーなどの筋疾患と関連しています。これらの疾患では、ジスフェルリンの欠損や機能不全により、筋膜の修復が適切に行われなくなり、筋細胞が徐々に損傷を受け、筋力低下や筋肉の喪失が進行します。これらの疾患では、患者が運動機能を徐々に失っていくことが特徴です。
● まとめ
ジスフェルリンは、筋細胞膜の修復において重要な役割を果たしており、筋膜の損傷が起こった際にその修復を促します。さらに、筋再生や炎症にも関与している可能性がありますが、これらの機能についてはまだ多くが不明です。DYSF遺伝子の変異が、筋疾患の発症と進行に関連しているため、このタンパク質の機能解明は、将来的な治療開発においても重要な課題となっています。
遺伝子と関係のある疾患
遺伝子の発現とクローニング
Bashirら(1998年)は、常染色体劣性肢帯型筋ジストロフィー2(LGMDR2; 253601)に関連する2番染色体上のYACおよびPACクローンを解析し、dysferlin部分cDNAをクローニングしました。このタンパク質は推定1,779アミノ酸から構成され、少なくとも2つのC2ドメインとC末端に膜貫通ドメインを持つことが示唆されました。ノーザンブロット解析では、ジスフェルリンの発現は骨格筋、心臓、胎盤で顕著であり、肝臓、肺、腎臓、膵臓では弱く検出されました。また、4kb未満の転写体が脳にも存在し、特に小脳と髄質で7kbの転写体が検出されました。ジスフェルリンの役割は筋ジストロフィーに関連しており、その名称は筋肉における機能とC.エレガンス(線虫)のタンパク質との相同性に由来しています。
Brittonら(2000年)は、DYSF遺伝子が2,080個のアミノ酸残基から構成されることを確認しました。さらに、DYSFは6つのC2ドメイン、C末端の膜貫通ドメイン、そしてカルシウム結合に関連する複数の保存された残基を持っており、これらは線虫のferlin、myoferlin(MYOF)、otoferlin(OTOF)との間で保存されています。
Pramonoら(2006年)は、成体の骨格筋から採取したトータルRNAの5′ RACE解析により、DYSF遺伝子のエクソン1の代替スプライスバリアントを同定し、このバリアントをDYSF-v1と名付けました。このバリアントは、2,081アミノ酸から成り、N末端のみがDYSFと異なることが特徴です。ノーザンブロット分析では、DYSF-v1転写体(7.5 kb)が骨格筋、心臓、脾臓、小腸、腎臓、肝臓、胎盤、肺で検出され、脳では6-kbの転写体が確認されました。また、2-kbや1.3-kbのマイナーな転写体も報告されました。
7.5-kbのDYSF転写体は、脳、心臓、骨格筋、脾臓、腎臓、肝臓、小腸、胎盤、肺、そして末梢血白血球で広く検出されており、他にも5-kb、2-kb、1.3-kbのマイナーな転写体がそれぞれ異なる組織で発現していました。DYSF-v1に関連するプロモーター領域には、2つのCpGアイランド、TATAボックス、および筋肉発現に関与する転写因子の結合部位を含む2つのクラスターが存在しました。
さらに、Pramonoらは、中国、マレーシア、インド出身の50人の血液サンプルと、無関係な個人の骨格筋サンプルを用いたRT-PCR解析により、DYSF遺伝子の新しい選択的スプライシングバリアントを特定しました。この中には、エクソン5aおよびエクソン40aといった新規エクソンが含まれており、さらに以前報告されたエクソン17の選択的スプライシングも確認されました。
長距離RT-PCRとサブクローニングにより、14種類のジスフェルリン転写体が同定され、これらすべてでオープンリーディングフレームが維持されていることが明らかになりました。また、骨格筋と血液における転写体の相対頻度に違いがあることも示されました。Pramonoら(2009年)は、これらの知見がDYSF関連疾患の分子診断において臨床的に関連する可能性があると指摘しています。
Pramonoらの研究は、DYSF遺伝子のスプライシングバリアントが豊富であり、その発現パターンが異なる組織において多様であることを明らかにしました。これらの知見は、DYSF関連疾患の分子診断や治療のための新たな視点を提供するものと考えられます。
遺伝子の構造
一方、Pramonoら(2006年)は、DYSF遺伝子の第1イントロン内に位置する代替第1エクソンに関連するプロモーター領域を特定し、このプロモーター領域が新しいスプライスバリアントDYSF-v1の発現に関与していることを示しました。DYSF-v1は、他のDYSF転写物と異なり、N末端の構造が異なっています。この発見は、ジスフェルリン遺伝子の発現制御の複雑さを示すものであり、選択的スプライシングがDYSFの機能多様性に寄与していることを示唆しています。
● まとめ
– Aokiら(2001年)は、DYSF遺伝子が55エクソンから成ることを明らかにしました。
– Pramonoら(2006年)は、DYSF遺伝子の第1イントロン内にある代替第1エクソンに関連するプロモーター領域を特定し、スプライスバリアントDYSF-v1の発現に関与していることを報告しました。
このような発見は、DYSF遺伝子の複雑なスプライシングメカニズムや機能多様性を理解するための重要な手がかりとなっています。
マッピング
遺伝子の機能
Matsudaら(2001年)は、カベオリン-3(CAV3)とジスフェルリンが共免疫沈降することを報告し、カベオリン-3は骨格筋膜タンパク質であり、肢帯型筋ジストロフィー(LGMD1C)やリップリング筋疾患(RMD2)に関連しています。免疫蛍光法を用いて、LGMD1C患者の筋肉ではジスフェルリンの異常な局在が確認されました。ジスフェルリンには、カベオリン-3の足場結合モチーフに対応する7つの部位と、カベオリン-3のWWドメインに結合する可能性のある部位があることが示されました。これにより、ジスフェルリンがカベオリン-3と相互作用し、カベオラのシグナル伝達機能を補助している可能性があるという仮説が提唱されました。
Fujitaら(2007年)は、C2C5マウス細胞において、ジスフェルリンがユビキチン/プロテアソームER関連分解系(ERAD)によって分解されることを発見しました。プロテアソーム阻害剤を使用すると、ジスフェルリンが小胞体に蓄積しましたが、変異型ジスフェルリン(例:W999C変異)は小胞体内で凝集し、ERストレスを引き起こしました。このストレスにより、EIF2-alphaのリン酸化を通じてオートファゴソームの形成が促進され、ERストレスによる細胞死も引き起こされました。リソソームプロテアーゼ阻害剤によって変異型ジスフェルリンの凝集体が蓄積することも示されました。この研究では、2つのERADモデルが提案されました。通常のERAD(I)ではユビキチン/プロテアソーム系が機能しますが、変異型ジスフェルリンが凝集すると、このシステムが損なわれ、ERAD(II)としてオートファジー/リソソーム経路が代替的に機能します。
● まとめ
– Andersonら(1999年)は、ジスフェルリンが筋繊維膜に局在し、DYSF遺伝子に変異がある患者ではジスフェルリンが欠如していることを発見しました。
– Matsudaら(2001年)は、ジスフェルリンとカベオリン-3が相互作用し、カベオラのシグナル伝達に関与している可能性を提唱しました。
– Fujitaら(2007年)は、ジスフェルリンがERストレス下で異常に凝集し、ERADシステムによって処理されることを示し、2つの分解経路を提案しました。
分子遺伝学
Weilerら(1996年)およびIllarioshkinら(1996年、1997年)は、MMD1とLGMD2Bの両方を示す患者が含まれる2つの大規模な近親婚家系を報告し、罹患した個人が同じハプロタイプを共有していることを示しました。表現型の違いは、遺伝的修飾因子や環境要因などの追加の要因による可能性が示唆されました。
Liuら(1998年)は、MMD1、LGMD2B、遠位筋症(DMAT; 606768)に関連するDYSF遺伝子の9つの変異を報告し、そのうちの5つがジスフェルリンの発現を妨げると予測しました。これにより、同じDYSF遺伝子変異が複数のミオパチー表現型を引き起こすことが確認されました。
Bashirら(1998年)は、DYSF遺伝子におけるホモ接合性フレームシフト変異(603009.0005および603009.0006)を特定し、近位および遠位筋ジストロフィーの両方を発症することを確認しました。
Matsumuraら(1999年)およびAokiら(2001年)は、三好ミオパチーの患者において、ジスフェルリン遺伝子に複数の変異(例:603009.0010および603009.0011)を特定しました。
Weilerら(1999年)は、同じ家系においてLGMD2Bと三好型ミオパチーが発生する問題を指摘し、同じ変異がこれらの異なる疾患を引き起こす可能性があることを示しました。この発見により、DYSF遺伝子の役割が異なる臨床的表現型に与える影響を理解するための基礎が確立されました。カナダの先住民家系では、すべての患者がジスフェルリンのプロリン791からアルギニンへの変異(603009.0007)をホモ接合型で持ち、同じ変異が異なる疾患を引き起こす原因となっていることが確認されました。
高橋氏らは、臨床的に三好ミオパチーと診断された日本人患者25人中20人において、16種類のジスフェルリン変異(そのうち10種類は新規)を特定しました。
Nguyen氏らは、ジスフェルリン症患者40人を対象にレビューを行い、約50%が典型的な三好ミオパチーまたはLGMD2Bを示し、その他の患者は混合型発症(35%)、足の腫れ(10%)、無症候性の血清クレアチンキナーゼ増加(5%)などのより珍しい表現型を示すことを明らかにしました。この疾患はしばしば急速に悪化し、25%の患者が最初に多発性筋炎と誤診されていました。
● まとめ
– 三好ミオパチー(MMD1)とLGMD2Bは、同じDYSF遺伝子変異に起因する異なる表現型であり、同一遺伝子の変異が複数の異なる筋疾患を引き起こすことが確認されています。
– 表現型の違いには、修飾因子や環境要因が関与している可能性が高いとされています。
遺伝子型と表現型の相関
著者らは、これらのアミロイド線維がジスフェルリンのタンパク質分解によって生成されたものであり、変異によってジスフェルリンの構造が不安定化し、その結果、アミロイド線維の形成が促進される傾向があると推測しました。通常、アミロイド線維は異常なタンパク質の凝集によって形成されるため、ジスフェルリンの変異がタンパク質の誤った折りたたみや凝集を引き起こし、それが筋疾患の一因となる可能性があります。
●まとめ
– Spulerら(2008年)は、DYSF遺伝子のN末端変異がジスフェルリンの構造を不安定化し、筋肉線維内にアミロイド線維が沈着することを報告しました。
– これらのアミロイド線維は、ジスフェルリンのタンパク質分解生成物であり、変異によってアミロイド線維を形成する傾向が高まると推測されています。
動物モデル
Bansalら(2003年)は、ジスフェルリンノックアウトマウスを作製し、このマウスが進行性筋ジストロフィーを発症することを発見しました。ジスフェルリンノックアウト筋線維では、筋細胞膜の損傷に応じた膜修復ができず、カルシウム依存性の膜修復に欠陥があることが明らかになりました。これにより、ジスフェルリンが筋細胞膜の修復プロセスに不可欠な役割を果たしていることが示されました。
Hoら(2004年)は、ジスフェルリン欠損マウスを作製し、これらのマウスが進行性筋ジストロフィーを発症することを確認しました。ジスフェルリン欠損マウスでは、筋細胞膜の完全性が失われ、ジストロフィン糖タンパク質複合体は保たれているものの、筋膜の破壊と筋線維の変性が観察されました。
Hanら(2007年)は、ジスフェルリンノックアウトマウスにおいて軽度の心筋症が発生し、ストレス運動により悪化することを報告しました。ジスフェルリンを介した膜修復が心筋細胞の膜の完全性を維持するために重要であることが示唆されました。
Chiuら(2009年)は、ジスフェルリン欠損マウスにおいて、筋再生の減弱や壊死線維の除去遅延、炎症期の延長が観察され、ジスフェルリンが化学走性分子の分泌に関与している可能性を示唆しました。
Gloverら(2010年)は、ヒトジスフェルリンを過剰発現する遺伝子導入マウスを作製し、ジスフェルリンの高レベルの過剰発現が筋萎縮や重度の後弯、筋力低下を引き起こすことを報告しました。この研究は、ジスフェルリンの遺伝子治療において、発現レベルや投与量が重要な要素であることを示唆しました。
● まとめ
– SJLマウスは自己免疫疾患や筋再生のモデルとして有用であり、ジスフェルリンの欠損が筋ジストロフィーの進行に関与しています。
– ジスフェルリンは筋細胞膜の修復に重要な役割を果たしており、その欠損は筋疾患や心筋症を引き起こします。
– ジスフェルリンの過剰発現もまた、筋疾患を引き起こす可能性があり、遺伝子治療において適切な発現レベルの調整が重要です。
アレリックバリアント
DYSF, GLN605TER
三好筋ジストロフィー(MMD1; 254130)のフランス人家族において、Liu ら(1998)は、DYSF 遺伝子のコドン605におけるホモ接合性ナンセンス変異、すなわちヌクレオチド2186におけるCAGからTAGへの転位(Q605X)が、罹患した家族メンバーに認められることを発見しました。
0.0002 前脛骨筋に発症する遠位型ミオパチー
DYSF、1-BP欠失、5966G
スペインの近親婚家族において、前脛骨筋に発症する遠位型ミオパチー(DMAT;606768)と診断されたメンバーがいたが、Liuら(1998年)は、このミオパチーがDYSF遺伝子における5966Gの欠失と関連しており、フレームシフトを引き起こしていることを発見した。Illa ら(2001)は、同じ家族についてさらに詳細に報告し、この疾患を別のジスフェルリノパチーと呼んでいます。この家族の罹患したメンバーは、欠失についてホモ接合でした。.
0003 三好型筋ジストロフィー
肢帯型筋ジストロフィー、常染色体劣性2、含まれる
DYSF, ILE1298VAL
三好型筋ジストロフィー(MMD1; 254130)患者と肢帯型筋ジストロフィー2B型(LGMDR2; 253601)患者がいるイタリアの家族において、すなわち、それぞれ遠位型または近位型ミオパチーであることが判明している家族において、Liu ら (1998) は DYSF遺伝子における2つのミスセンス変異の複合ヘテロ接合性を発見しました。4265ヌクレオチドにおけるATGからGTCへの転位(I1298V)と、6497ヌクレオチドにおけるCGTからTGTへの転位(R2042C; 603009.0004)です。
.0004 三好型筋ジストロフィー
肢帯型筋ジストロフィー、常染色体劣性2、
DYSF、ARG2042CYS
三好筋ジストロフィー(MMD1; 25413 。
肢帯型筋ジストロフィー2B型(LGMDR2; 253601)の患者で複合ヘテロ接合状態で発見されたDYSF遺伝子の2042-to-cys(R2042C)変異については、Liuら(1998年)の文献603009.0003. .
0005を参照のこと。 肢帯型筋ジストロフィー、常染色体劣性2
DYSF、5-bp欠失/4-bp挿入、NT4872
肢帯型筋ジストロフィー2B型(LGMDR2; 253601)の8つのリビア系ユダヤ人家族において、Bashir ら(1998)は、罹患した個体がグアニンの1bp欠失と DYSF遺伝子のコドン1322におけるグアニンの1塩基欠失とC-G転換のホモ接合型であり、その結果、1331番目の位置でフレームシフトと早期終止コドンが生じることが分かりました(DYSFアミノ酸配列の一部に基づく番号付け)。その後、Therrienら(2006年)は、この変異を挿入/欠失(4872delinsCCCC)として報告しました。リビア系ユダヤ人の9家族のうち、患者が1人いる家族では、変異は1コピーで検出されました。両親の1人(変異は保有していない)はルーマニア出身でした。これらの家族における25人の患者は、12歳から39歳(平均19.5±5歳)で発症しました。全員が、上肢症状の平均9年前に下肢症状を発症していました。13人の患者(52%)は遠位の下肢筋の筋力低下を示し、そのほとんどは腓腹筋で、一時的なふくらはぎの腫れを訴える患者もいました。 筋力低下の分布には家族内でのばらつきが見られました。 6人の患者のみが自立歩行能力を失っており、その全員が35歳以上でした。 筋生検では慢性の筋疾患の変化が認められ、クレアチンキナーゼはすべての患者で正常値の10~25倍に上昇していました。
Sinnreich 氏らは、重度の LGMDR2 を患うイタリア人姉妹2人において、DYSF 遺伝子における挿入/欠失変異のホモ接合性を確認しました。この変異は、Bashir 氏ら(1998年)によって以前に報告された1bp欠失(4872delG)および4876G-C転換であると特徴づけられました。両親はそれぞれ、挿入/欠失変異のヘテロ接合型でした。 少女たちは、近位筋の脱力および筋力低下が10代で発症しました。 70歳の母親は、40代から軽度の近位筋脱力があったため、indel変異とスプライス部位変異の複合ヘテロ接合型でした(603009.0016)。
.0006 肢帯型筋ジストロフィー2
DYSF、23-BP INS
Mahjneh ら(1992年)が報告したパレスチナ系アラブ人家族において、Bashir ら(1998年)は、常染色体劣性遺伝の肢帯型筋ジストロフィー2B型(LGMDR2; 253601)が、DYSF遺伝子のコドン1386における23塩基対の挿入によって生じていることを明らかにしました。この挿入は、複製ずれによるタンデム重複であり、コドン1427でフレームシフトと早期終結が起こると予測されました(DYSFアミノ酸配列の一部に基づく番号付け)。この近親者には先天性筋ジストロフィー(254300参照)の患者も含まれていましたが、これらの患者のいずれもDYSF変異は保有しておらず、異なる遺伝的基盤を持つことが示されました。
0.0007 肢帯型筋ジストロフィー、常染色体劣性 2
三好型筋ジストロフィー 1、
DYSF、PRO791ARG
Weilerら(1996年)が報告したカナダの先住民の大家族において、Weilerら(1999年)は、肢帯型筋ジストロフィー2B型(LGMDR2; 2 53601)と三好型筋ジストロフィー(MMD1; 254130)患者の両方が、DYSF遺伝子におけるプロ791からアルギニン(P791R)へのミスセンス変異のホモ接合型であることが分かりました。 さらに、未発表の2つの家族から4人の患者が、この変異を有していました。 ハプロタイプ分析により、すべての患者における変異の共通の起源が示唆されました。三好型ミオパチーまたはLGMD2Bの患者の筋組織のウェスタンブロット分析では、それぞれ15%と11%の同程度の豊富なジスフェルリン染色が認められました。正常な組織切片では、ジスフェルリンが筋細胞膜に局在していることが示されましたが、三好型ミオパチーまたは肢帯型筋ジストロフィーの患者の組織切片では、2つのタイプを区別できないほど染色がほとんど認められませんでした。
0.0008 常染色体劣性肢帯型筋ジストロフィー2
DYSF、NT5711、G-A、+5
イエメン系ユダヤ人の肢帯型筋ジストロフィー2B型(LGMDR2; 253601)の近親交配家系において、McNallyら(2000年)は、ジスフェルリンcDNA配列のアミノ酸1686番目(5711bp)に続くイントロン5番目の位置に影響を与えると予測されるホモ接合型G-to-A変異を特定しました。この突然変異に関連する表現型は、20歳代後半から始まり、血清クレアチンキナーゼの上昇が含まれます。これらの患者の生検サンプルでは、炎症過程が認められましたが、これは以前は肢帯型筋ジストロフィーとは関連付けられていませんでした。
0.0009 肢帯型筋ジストロフィー、常染色体劣性 2
三好型筋ジストロフィー 1、
Illarioshkin ら (1996) により肢帯型筋ジストロフィー 2B 型 (LGMDR2; 253601) および三好型ミオパチー (MMD1; 254130) の両方を持つ近親ロシア人大家族が報告されましたが、 、Illarioshkin ら(2000年)は、すべての患者がDYSF遺伝子のヌクレオチド573-574におけるTGからATへの変異についてホモ接合であり、バリン67からアスパラギン酸(V67D)への置換が生じていることを発見しました。 .
0010 三好型筋ジストロフィー1型
DYSF, TRP999CYS
三好筋ジストロフィー(MMD1; 254130)患者において、松村ら(1999年)はDYSF遺伝子のエクソン28における3370G-T転換を特定し、その結果、トリプトファン999がシステイン(W999C)に置換されました。高橋ら(2003)は、一般的に、W999C 変異を持つ患者は発症年齢が遅く(平均32歳)、症状も軽度である傾向があることを指摘しています。 .
0011 三好型筋ジストロフィー1
DYSF、ARG1046HIS
三好筋ジストロフィー(MMD1; 254130)患者において、Aoki ら(2001)はDYSF遺伝子のエクソン29における3510G-A転位を特定し、アルギニン1046がヒスチジン(R1046H)に置換されることを明らかにしました。高橋ら(2003年)は、R1046H変異を持つ患者は発症年齢が比較的若く(平均13歳)、DYSF遺伝子の他の変異を持つ患者よりも有意に高い血清クレアチンキナーゼ値を示す重症型であることを指摘しています。
0.0012 三好型筋ジストロフィー
前脛骨筋遠位型ミオパチー(脛骨前面発症)
肢帯型筋ジストロフィー(常染色体劣性遺伝)2
ジスフェルリノパチー、アルギニン1905番目の終止
スペインのSueca出身の5家族の筋ジストロフィー患者において、Vilchezら(2005年)は、DYSF遺伝子のエクソン51におけるホモ接合型6086C-T転位を特定し、その結果、arg1905-to-ter(R1905X)置換が生じました。2つの家族は三好型ミオパチー(MMD1; 254130)を発症し、2つの家族は前脛骨筋を始まりとする遠位筋ミオパチー(DMAT; 606768)を発症し、1つの家族は肢帯型筋ジストロフィー2B型(LGMDR2; 253601)を発症しました。同じ突然変異が異なる診断結果をもたらしたものの、各家族の患者は同じ表現型を示しました。R1905X 突然変異は、168本の対照染色体では確認されませんでした。ハプロタイプ分析により、創始者効果があることが示されました。Suecaは1245年に、アラゴン王ハイメ1世からバレンシアのムーア人からの再征服の功績として土地を授けられた病院修道会に属する17人の入植者によって創設されました。
0013 肢帯型筋ジストロフィー、常染色体劣性 2
DYSF、ASP625TYR
古典的肢帯型筋ジストロフィー2B型(LGMDR2; 253601)の男性において、Illa ら(2007年)は、DYSF遺伝子における2つの変異の複合ヘテロ接合性を特定しました。エクソン2の1873G-T転換により 0エクソンにおける1873G-Tトランスバージョンにより、asp625がtyr(D625Y)に置換し、エクソン47における5201A-G転位により、glu1734がgly(E1734G; 603009.0014)に置換しました。患者の54歳の姉は、D625Y変異のヘテロ接合型であり、51歳時に歩行中の進行性疲労と階段昇降困難を発症しました。彼女は下肢近位筋の筋力低下、血清クレアチンキナーゼの増加、およびMRIで下肢筋の脂肪浸潤の証拠が認められました。免疫染色およびウェスタンブロット分析では、この女性の筋におけるジスフェルリンレベルの低下が示されましたが、RT-PCRではDYSF mRNAのレベルは正常でした。これらの所見は、ヘテロ接合型DYSF変異キャリアが、遅発性の軽度の症状を発症する可能性を示唆しています。
0.0014 肢帯型筋ジストロフィー、常染色体劣性 2
DYSF、GLU1734GLY
Illa ら(2007年)により肢帯型筋ジストロフィー2B型(LGMDR2; 253601)患者において複合ヘテロ接合状態で発見されたDYSF遺伝子のグルタミン酸1734-グリシン(E1734G)変異に関する考察については、603009.0013を参照してください。
.0015 三好筋ジストロフィー1型
DYSF、GLY519ARG
Illa ら(2007年)は、三好筋ジストロフィー(MMD1;254130)の2人の同胞において、DYSF遺伝子のエクソン18における1555G-A転位のホモ接合体を同定し、グリシン519がアルギニン(G519R)に置換していることを明らかにしました。発症年齢はそれぞれ18歳と15歳で、遠位の下肢筋力低下から始まり、近位筋、後に上肢筋にも障害が及ぶようになりました。 両名とも30代で車椅子生活を余儀なくされました。 G519R変異のヘテロ接合型であった患者の父親は、65歳でふくらはぎの筋肉痛と歩行困難を訴え、徐々に進行しました。血清クレアチンキナーゼの値が中程度に上昇し、筋生検ではジスフェルリンの免疫染色が低下していましたが、DYSF mRNA レベルは正常でした。 この所見は、ヘテロ接合型 DYSF 変異保有者が、遅発性の軽度の症状を発症する可能性を示唆しています。
0.0016 肢帯型筋ジストロフィー、常染色体劣性 2
DYSF、IVS31DS、A-G、-33
軽度の肢帯型筋ジストロフィー2B型(LGMDR2; 253601)の女性において、Sinnreich ら(2006年)は、 DYSF遺伝子における2つの変異、すなわち、エクソン31におけるA-to-G転位によるエクソン32のインフレームスキップ、およびインデル変異(603009.0005)が複合ヘテロ接合性であることを同定しました。A-to-G変異は、遺伝子内の2つの推定ラリアット分岐点配列のうちの2番目の配列に位置することが分かりました。ウェスタンブロット分析では、ジスフェルリンのレベルが正常の約10%に減少していることが示されました。患者の2人の重度の影響を受けた娘は、indel変異のホモ接合型でした。 Sinnreich ら (2006) は、母親の軽度の表現型はスプライス部位の変異によるタンパク質の機能の残存によるものだと仮定しました。
0.0017 肢帯型筋ジストロフィー、常染色体劣性 2
DYSF, GLY299ARG
肢帯型筋ジストロフィー2B型(LGMDR2; 253601)の2人の同胞において、Spulerら(2008年)はDYSF遺伝子における2つの変異について複合ヘテロ接合性を特定しました。895G-A転位により 2番目のC2ドメインの下流でgly299-to-arg(G299R)置換を引き起こす895G-A転移、およびナンセンス媒介分解とタンパク質発現の欠如を引き起こすことが予測される1bp欠失(855+1delG; 603009.0020)が同定されました。両患者の骨格筋生検では、ジスフェルリンの2番目のC2ドメインに対する抗体で染色されたアミロイド線維が認められました。アミロイドは筋細胞の筋膜、多数の血管壁、および間質に存在していました。 発症していない父親はG299R変異のヘテロ接合型であり、骨格筋にはアミロイドは認められませんでした。 Spulerら(2008年)は、この変異によりタンパク質の構造が不安定になり、アミロイド線維を形成する傾向が高まったと推測しています。このコドンにおける別の変異(G299W;603009.0018)を有する筋疾患の無関係な家系でも、骨格筋線維にアミロイドーシスが認められました。
0018 三好筋ジストロフィー1型
DYSF、GLY299TRP
ミオシス(MMD1; 254130)の2人の同胞において、Spulerら(2008年)はDYSF遺伝子におけるホモ接合型895G-T転換を同定し、その結果、2番目のC2ドメインの下流でグリシン299がトリプトファン(G299W)に置換しました。1人の患者から採取した骨格筋生検では、ジスフェルリンの第2 C2 ドメインに対する抗体で染色されたアミロイド線維が認められました。アミロイドは筋細胞の筋膜、多数の血管壁、および間質に存在していました。Spuler ら(2008年)は、この変異によりタンパク質の構造が不安定になり、アミロイド線維が形成されやすくなったと推測しています。ミオパチーを患う無関係な家族で、このコドンにおける別の変異(G299R; 603009.0017)も、骨格筋線維におけるアミロイドーシスを示しました。.
0019 肢帯型筋ジストロフィー、常染色体劣性 2
DYSF、IVS14AS、A-G、-2
肢帯型筋ジストロフィー2B型(LGMDR2; 253601)のアラブ人男性において、Spuler ら(2008年)は、DYSF 遺伝子のイントロン14におけるホモ接合型A-to-G転位を同定し、その結果、第3のC2ドメインの近傍にスプライス部位の変異が生じました。筋生検では、筋細胞膜の欠陥とアミロイド線維の沈着が認められました。Spuler ら(2008)は、この変異がタンパク質の構造を不安定にし、アミロイド線維を形成する傾向を高めると推定しています。
0.0020 肢帯型筋ジストロフィー、常染色体劣性 2
DYSF、1-bp欠失、855+1G
Spuler ら(2008 年)により肢帯型筋ジストロフィー 2B 型(LGMDR2; 253601)患者において複合ヘテロ接合状態で発見された DYSF 遺伝子(855+1delG)の 1bp 欠失の議論については、603009.0017 を参照してください。
.0021 肢帯型筋ジストロフィー、常染色体劣性 2
DYSF、1-BP 欠失、2776G
2歳と5歳のスペイン人の同胞において、肢帯型筋ジストロフィー2B型(LGMDR2; 253601)の先天性発症という珍しい症例について、Paradas et al. (2009) はDYSF遺伝子のエクソン26におけるホモ接合型1bp欠失(2776delG)を同定しました。その結果、フレームシフトと早期終結が起こりました。両親は血族関係にはありませんでしたが、同じ小さな村の出身であり、ハプロタイプ分析では古代の血族関係が示唆されました。 2人の患者はともに乳児期に低緊張と運動発達の遅れで発症しました。 歩行、走行、階段昇降が困難で、頚部筋力低下も見られました。患者の筋肉ではジスフェルリンの発現がほとんど認められなかったのに対し、この突然変異のヘテロ接合体の臨床症状を示さない家族のメンバーでは、ジスフェルリンの発現が約50%減少していました。Paradas ら(2009年)は、この兄弟姉妹におけるこの疾患の早期発症を強調し、彼らはDYSF突然変異と関連付けられていなかった新しい表現型を有している可能性があると示唆しています。
0.0022 肢帯型筋ジストロフィー、常染色体劣性 2
DYSF、5492G-A
肢帯型筋ジストロフィー2B型(LGMDR2; 253601)のポルトガル人男性患者6人のうち、無関係の患者において、サントス氏ら(2010年)は、DYSF遺伝子のエクソン49の最後のヌクレオチドにおけるホモ接合型5492G-A転位を特定しました。別のポルトガル人患者は、この変異とDYSF遺伝子の別の病原性変異との複合ヘテロ接合型でした。5492G-A変異は、240の対照アレルでは認められませんでした。患者の組織の転写解析により、5492G-A変異が異常スプライシング、エクソン49のスキップ、早期終結を引き起こしていることが示されました。さらに研究を進めたところ、患者と正常対照者(特に血液)の両方において、エクソン50と51が関与する選択的スプライシングのいくつかの残存発現産物が特定されました。ハプロタイプ解析により、この変異が創始者効果によるものであることが裏付けられました。7人の患者は全員、ポルトガル北部の内陸部の限られた地域に起源または居住していました。ほとんどの患者は肢帯型筋ジストロフィーでしたが、いくつかの表現型のばらつきがありました。1人の患者は下肢遠位筋の筋力低下を示し、別の患者は心不整脈を発症しました。







