承認済シンボル:DNAI1
遺伝子名:dynein axonemal intermediate chain 1
参照:
HGNC: 2954
AllianceGenome : HGNC : 2954
NCBI:27019
Ensembl :ENSG00000122735
UCSC : DNAI1 (ENST00000614641.4) from GENCODE V46
遺伝子OMIM番号604366
●遺伝子のlocus type :タンパク質をコードする
●遺伝子のグループ:Dyneins, axonemal outer arm complex subunits
WD repeat domain containing
●遺伝子座: 9p13.3
●ゲノム座標:9:34,458,805-34,520,984
遺伝子の別名
CILD1
DIC1
DNAI1_HUMAN
dynein intermediate chain 1, axonemal
dynein intermediate chain DNAI1
dynein, axonemal, intermediate chain 1
dynein, axonemal, intermediate polypeptide 1
IC78
ICS1
immotile cilia syndrome 1
MGC26204
遺伝子の概要
ダイニンは、軸糸内で「内側ダイニン腕(IDA)」と「外側ダイニン腕(ODA)」という構造の一部を形成します。DNAI1遺伝子が作る中間鎖1は、ODAに含まれ、繊毛の運動に必要な力を発生させる重要な要素です。ダイニン複合体は、重鎖、中間鎖、軽鎖という異なるタンパク質サブユニットから成り、それぞれが異なる遺伝子によって生成されます。DNAI1遺伝子は、ODAの構造や機能に特に重要な役割を果たしているため、DNAI1遺伝子に変異があると、繊毛の動きに異常が生じ、原発性線毛機能不全症(PCD)などの疾患につながることがあります。
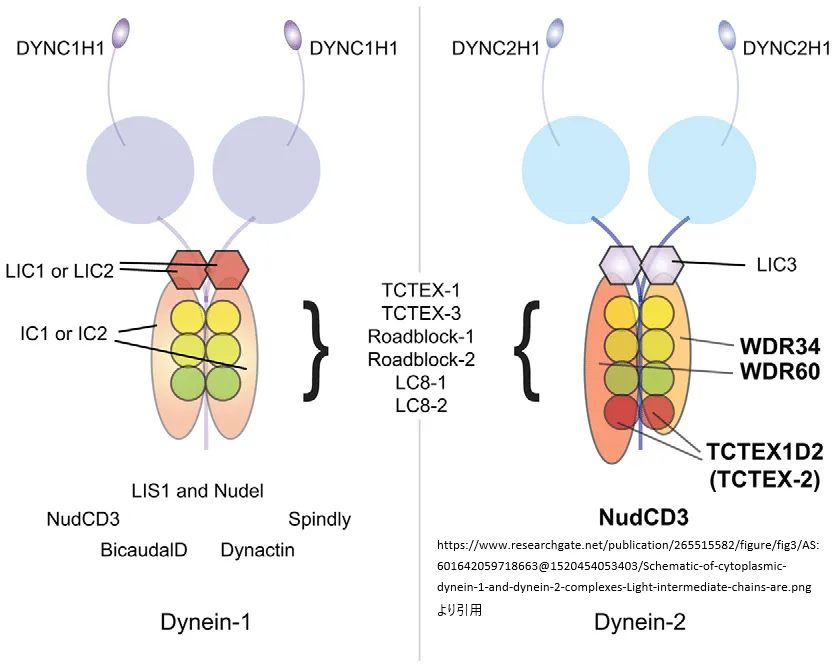
www.researchgate.net/publication/265515582/figure/fig3/AS:601642059718663@1520454053403/Schematic-of-cytoplasmic-dynein-1-and-dynein-2-complexes-Light-intermediate-chains-are.png
より引用
細胞質ダイニン-1およびダイニン-2複合体の模式図。軽鎖は六角形で、中間鎖は細長い楕円形で、軽鎖は円形で示されています。ダイニン-1(左)には、ダイニン-2(右)には見られない追加の相互作用パートナーが存在しています。NudCD3は、両方の細胞質ダイニン複合体と結合しています。TCTEX1D2が複合体内で単量体または二量体のいずれで存在しているかは、依然として不明です。
遺伝子と関係のある疾患
遺伝子の発現とクローニング
ダイニンアームに関する詳細な構造や生化学的研究は、クラミドモナス・レヒンダイト(Chlamydomonas reinhardtii)などの単細胞緑藻で盛んに行われています。クラミドモナスは、ヒトの呼吸繊毛や精子尾部と類似した「軸糸」構造を持つ鞭毛を持ち、これを用いて運動するため、原発性線毛機能不全症(PCD)研究のモデルとして適しています。特に、クラミドモナスの鞭毛におけるダイニンアームの欠陥は、PCD患者の繊毛や精子尾部に見られる超微細構造異常と非常に類似していることが多いです。
クラミドモナスの鞭毛において、IC78と呼ばれる中間鎖(インターメディエイトチェーン)に欠陥がある突然変異体は、原発性線毛機能不全症と同様の軸糸異常を示すため、詳細な機能解析や分子レベルの研究が行われてきました。外ダイニンアームの欠如は、PCD患者でよく見られる特徴的な異常であり、この点でIC78欠損のクラミドモナス変異体は特に注目されています【244400】。
Pennarunら(1999年)は、クラミドモナスIC78遺伝子の研究に基づき、進化的に保存されているヒト遺伝子であるDNAI1遺伝子を単離し、PCDとの関連を調べました。DNAI1遺伝子は、699アミノ酸からなるタンパク質をコードしており、このタンパク質が外ダイニンアームの中間鎖の一部を構成しています。ノーザンブロット分析では、この遺伝子の転写産物が特に気管と精巣で豊富に発現していることが確認されており、繊毛や精子の運動機能において重要な役割を果たしていることが示唆されています。
遺伝子の構造
マッピング
その後、Gross (2013) は、DNAI1の配列情報(GenBank AF091619)とゲノム配列(GRCh37)のアラインメントに基づき、より正確に9p13.3染色体にDNAI1遺伝子をマッピングしました。これにより、DNAI1の位置が細かく特定され、関連する疾患の研究においてより正確な遺伝子解析が可能になりました。
分子遺伝学
アレリックバリアント
DNAI1、IVS1DS、1-BP INS、+3T
逆位を伴わない不動線毛症候群(244400)の9歳男児において、Pennarun ら(1999年)はDNAI1遺伝子における2つの変異について複合ヘテロ接合性を発見しました。(T)はエクソン1に続くイントロン・スプライス供与配列のヌクレオチド+3に生じたもので、父親から遺伝したものであり、4-bp挿入(AATA)はコドン95(604366.0002)に生じたもので、母親から遺伝したものです。父親由来の挿入は HpaI 部位の導入をもたらし、母親由来の挿入はフレームシフトを引き起こし、24アミノ酸下流の早期終止コドンにつながりました。.
.0002 原発性線毛機能不全症、1
DNAI1、4-BP 挿入、コドン 95
Pennarun ら(1999 年)により不動線毛症候群(244400)患者において複合ヘテロ接合状態で発見された DNAI1 遺伝子(AATA)のコドン 95 における 4bp 挿入変異については、604366.0001 を参照してください。
.0003 原発性線毛機能不全症、1
DNAI1、GLY515SER
カルタゲナー症候群(244400参照)の患者において、Guichard ら(2001)は、DNAI1 遺伝子のイントロン1における1bpの挿入(604366.0001)と、DNAI1 遺伝子のエクソン16における1543G-A 変異に起因するgly515-to-ser 突然変異の複合ヘテロ接合性を発見しました (604366.0001)と、DNAI1遺伝子のエクソン16における1543G-A転移に起因するgly515-to-ser変異が認められました。発端者の兄弟は、逆位を伴わない再発性の上下気道感染症と不妊症を患っていました。繰り返し実施された精子検査では、運動性のない精子の鞭毛が認められました。また、兄弟はともに尿管結石を患っており、これは、原発性線毛機能不全症やカルタゲナー症候群の患者では通常報告されない異常です。兄弟の線毛は、外ダイニンアームが欠損または短縮していました。
.0004 一次線毛機能不全症、1
DNAI1、12-BP欠失
Guichard ら (2001) は、カルタゲナー症候群(244400参照)の患者で、全内臓逆位、慢性副鼻腔炎、気管支炎、反復性中耳炎、および前頭洞の欠損症を患っている症例を報告しています。彼女は、一般的なスプライシング欠損(604366.0001)とDNAI1遺伝子の12bp欠失による複合ヘテロ接合型であり、その結果、WDドメイン4番目のアミノ酸が4つ分短縮されたタンパク質が生成されました。
| 遺伝子 | DNAI1 |
| 疾患名 | 原発性線毛機能不全症候群(DNAI1連関) |
| スーパーNIPTジーンプラスで検査対象のバリアント | c.48+2dupT c.1612G>A c.2001+1G>A c.1490G>A c.1569+1G>T c.1644G>A |
| 検出率 | >67% >95% |
| 分布 | 一般人口(世界中のどこにでもある普遍的な人口) アシュケナージ系ユダヤ |
| 引用 | Zariwala M.A. et al. (2006) Fedick A.M. et al. (2015) |
| 程度 | 中等度 |
| 遺伝形式 | 常染色体劣性 |
| 症状:引用元 | www.shouman.jp/disease/details/03_05_007/ |
| 症状 | 気道上皮細胞の表面には多数の線毛が存在し、線毛が連動して口側に向かって鞭を打つように運動することで、粘液層の流れが生じ、微粒子状の異物、細菌、喀痰などを排除して気道内を清浄に保つようになっている。この線毛が正常な構造と機能を発揮するためには、多数の遺伝子が関与していることが判明している。先天的な異常によって線毛運動が障害され、中耳、耳管、鼻、副鼻腔、咽頭を含めた呼吸器系の易感染性を呈する疾患を線毛機能不全症候群という。慢性副鼻腔炎や気管支拡張症を高頻度に合併する。 |
| 頻度 | 1万人~4万人に1人 |
| 保因者頻度 | |
| 新生児マススクリーニング |
↑↑↑↑↑↑ テーブルこの上にペースト ↑↑↑↑↑↑







