承認済シンボル:DMD
遺伝子名:dystrophin
参照:
HGNC: 2928
AllianceGenome : HGNC : 2928
NCBI:1756
Ensembl :ENSG00000198947
UCSC : DMD (ENST00000357033.9) from GENCODE V46
遺伝子OMIM番号300377
●遺伝子のlocus type :タンパク質をコードする
●遺伝子のグループ:
●遺伝子座: Xp21.2-p21.1
●ゲノム座標:X:31,119,222-33,339,388
遺伝子の別名
dystrophin (muscular dystrophy, Duchenne and Becker types), includes DXS142, DXS164, DXS206, DXS230, DXS239, DXS268, DXS269, DXS270, DXS272
mental retardation, X-linked 85
BMD
DXS142
DXS164
DXS206
DXS230
DXS239
DXS268
DXS269
DXS270
DXS272
muscular dystrophy, Duchenne and Becker types
遺伝子の概要
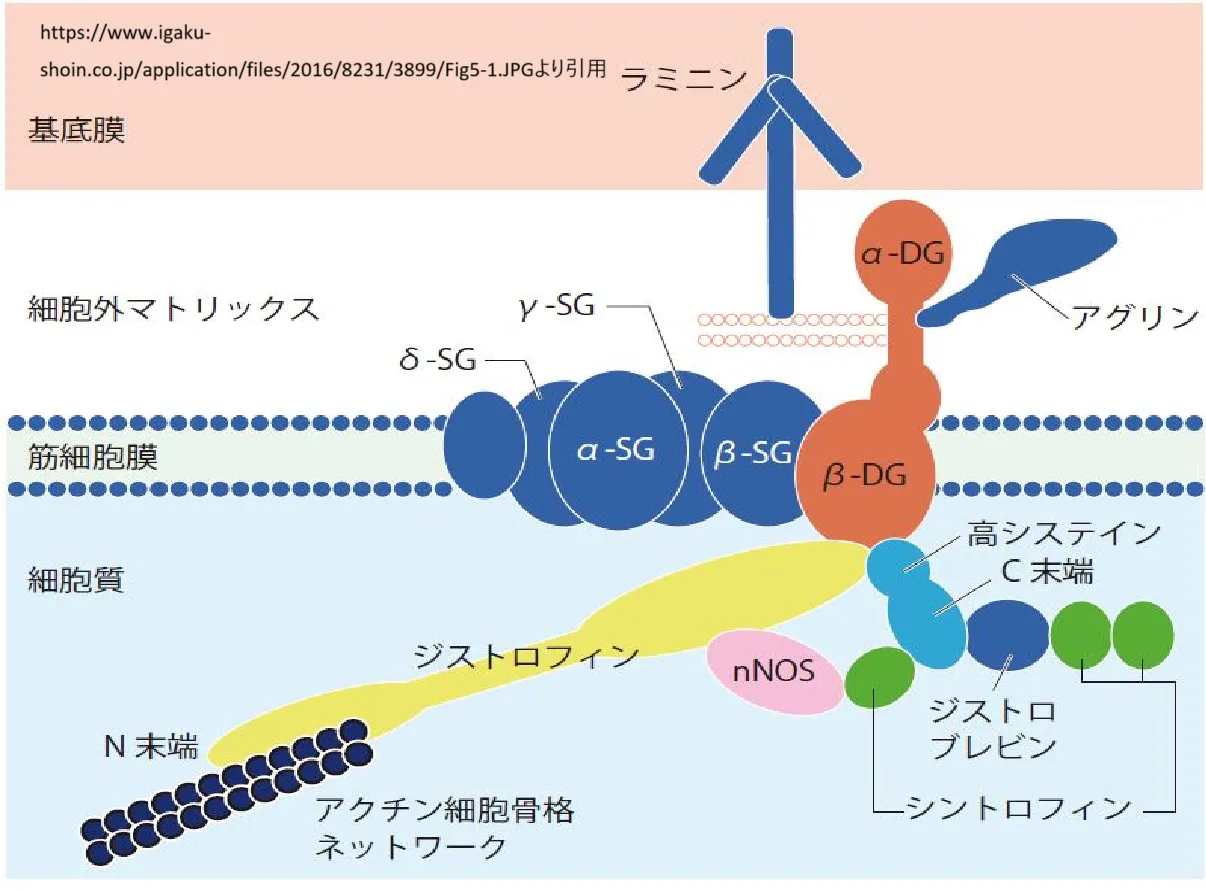
● 骨格筋と心筋におけるジストロフィンの機能:
– ジストロフィンは、筋肉の構造的枠組み(細胞骨格)と、細胞外のタンパク質を含む細胞外マトリックスをつなぐアンカーとして機能します。
– 筋肉が収縮や弛緩を繰り返すとき、ジストロフィン複合体は筋繊維を強化し、損傷から保護します。
– ジストロフィン複合体は、細胞シグナル伝達にも関与し、細胞間の化学信号のやり取りをサポートする可能性があります。
● 神経細胞におけるジストロフィンの役割:
– 神経細胞におけるジストロフィンの具体的な機能はまだ完全には解明されていませんが、研究によると、神経細胞間で情報をやり取りするシナプスの正常な構造や機能に重要であることが示されています。
● まとめ
ジストロフィンは、筋肉の機能を維持し、筋繊維を損傷から守るために不可欠なタンパク質であり、また、神経細胞間の通信にも役割を果たしていると考えられています。DMD遺伝子の異常が筋ジストロフィーなどの疾患の原因となることが知られています。
遺伝子と関係のある疾患
遺伝子の発現とクローニング
●ジストロフィン遺伝子とタンパク質の発見
Woodら(1987年)は、DMD遺伝子が約16kbのmRNAをコードしていることを示し、このmRNAが約500kDのタンパク質をコードすると予測しました。当時、筋肉内で知られていた最大のタンパク質は、ネブリリン(550kD)とティチン(1,000kD超)で、これらは筋肉のサルコメア構造に関与しています。DMD患者の筋肉には、ネブリリンに関連するバンドが欠如していることが確認されましたが、これは後に誤りであることが判明しました。
●ジストロフィンとαアクチニンの関係
その後の研究では、ジストロフィンがネブリリンではないことが明確になり、Koenig(1987年)は、DMD遺伝子がαアクチニンに似たタンパク質をコードしている可能性を示唆しました。αアクチニンは、筋肉のアクチンフィラメントと細胞骨格を結びつける重要なタンパク質であり、ジストロフィンも同様に、筋肉細胞膜を安定化させる役割を果たしていると考えられました。
●DMD遺伝子の解析
Hoffmanら(1987年)の研究では、ヒトおよびマウスのDMD遺伝子が約14kbの転写産物を持ち、アミノ酸配列がほぼ90%の相同性を示すことが明らかにされました。このことから、ジストロフィンが筋肉の構造的役割を担っている可能性が高いとされました。DMD遺伝子がコードするタンパク質は、約427kDの大きさを持つことが確認され、骨格筋や心筋、さらには胃の平滑筋でも見られました。
●ジストロフィンの構造と機能
Koenigら(1988年)は、ジストロフィンの全長配列を解読し、このタンパク質が3,685アミノ酸からなり、スペクトリンやαアクチニンと多くの共通点を持つことを発見しました。ジストロフィンは、筋肉細胞膜の強度を保つだけでなく、細胞骨格と細胞外マトリックスをつなぐ足場としても機能します。
●Hoffmanら(1987年)は、マウスcDNA由来の融合タンパク質に対するポリクローナル抗体を使用し、ヒトおよびマウスのDMD遺伝子がコードするタンパク質を特定しました。このタンパク質はジストロフィンと呼ばれ、約400kDの大きさで、横紋筋タンパク質の約0.002%を占めています。ジストロフィンは、骨格筋の三叉接合部と関連し、筋肉のカルシウムイオンの恒常性に関与している可能性が示唆されています。また、ジストロフィンは胃の平滑筋でも検出されており、筋型および脳型のジストロフィンアイソフォームはいずれも14kbのmRNAから翻訳され、427kDのタンパク質になります。
Koenigら(1988年)は、ジストロフィンのcDNA完全配列を報告し、このタンパク質が3,685アミノ酸から構成されていることを明らかにしました。ジストロフィンは、スペクトリンやαアクチニンと多くの構造的特徴を共有しており、長さ約150nmの棒状タンパク質であると予測されています。
●ジストロフィンのC末端と選択的スプライシング
ジストロフィンのC末端は、高度に保存された選択的スプライシング領域によってコードされています。Biesら(1992年)は、小児期発症のDMD患者において、1,824bpのゲノム内部3’領域の欠失を特定しました。この欠失は、システインに富んだCOOH末端ドメインが正確に切除されており、この領域がジストロフィンの重要な機能に関与していることが示唆されました。免疫細胞化学的分析により、ジストロフィンの染色は筋細胞膜に認められ、欠失が筋肉の機能に影響を与えることが確認されました。
●Zubrzycka-Gaarnら(1988年)の発見
Zubrzycka-Gaarnら(1988年)は、ヒトジストロフィンのN末端領域に由来する合成ペプチドと融合タンパク質に対する抗体を使用し、正常なヒト筋生検でジストロフィンが筋細胞膜に存在することを確認しました。この免疫反応性は、DMD患者では欠如または減少していましたが、他の筋疾患患者では正常なレベルが保たれていました。このことから、ジストロフィンは細胞骨格の要素を細胞膜に固定する役割を果たし、筋細胞膜を強化していると考えられます。
●Worton(1994年)は、ジストロフィンが筋肉細胞膜に単に機械的な強度を与えるだけでなく、より複雑な生物学的役割を果たしている可能性があると指摘しています。例えば、ジストロフィンは糖タンパク質複合体の特定の空間的配置を維持する役割を担っているかもしれません。ジストロフィンのアミノ酸配列は、スペクトリンや他の細胞骨格タンパク質と似ており、両端に球状ドメインを持つIビームのような構造をしています。
ジストロフィンは、次の2つの領域で異なる役割を果たします:
1. NH2末端は、細胞質内のアクチン線維と結合(筋肉の収縮を司るアクチンとは異なる)し、細胞骨格とのつながりを維持します。
2. C末端は、筋肉細胞膜に存在するジストロフィン結合タンパク質(DAP)やジストロフィン結合糖タンパク質(DAG)と結合します。この複合体は、筋細胞膜の安定性を保つために重要です。
これらのDAPやDAGは、分子量に基づいて次のように分類されています:
• 25DAP、35DAG、43DAG、50DAG、59DAP、156DAG
59DAPは細胞内に存在し、ジストロフィンに直接結合しているようです。一方、156DAG(ジストログリカン、DAG1としても知られています)は、細胞外マトリックスの主要成分であるラミニンと結合しています。これにより、細胞内外の構造的つながりが維持されます。
●ジストロフィンのC末端とβジストログリカンとの結合
Ishikawa-Sakuraiら(2004年)は、ジストロフィンのC末端領域に焦点を当て、βジストログリカンとの結合がZZドメインを介して行われていることを示しました。このドメインは、アミノ酸3026-3345にわたるジストロフィン断片に存在し、EF1ドメインと結合することで、βジストログリカンとの結合が強化されます。さらに、システイン残基3340は、ジストロフィンとβジストログリカンの結合に不可欠であり、この残基の変異(C3340Y)があると、ジストロフィンとβジストログリカンの結合が解離し、筋細胞膜でのジストロフィンの固定ができなくなることが示唆されています。
ユートロフィンとの違い
ユートロフィン(128240)は、ジストロフィンと類似の機能を持つタンパク質ですが、そのZZドメインに存在するシステイン残基の構造と結合様式は、ジストロフィンとは異なっており、それぞれが異なる機能を持つ可能性があります。
●臨床的および分子的意義
デュシャンヌ型およびベッカー型筋ジストロフィーは、ジストロフィン欠損または異常によって引き起こされる遺伝性筋疾患です。ジストロフィンの欠如は、筋繊維の脆弱性を増し、筋肉の収縮や弛緩時に損傷を受けやすくなります。この発見は、筋ジストロフィーに関する遺伝子治療や分子治療の研究に大きく貢献しています。
非筋ジストロフィン
チェリーら(1988年、1989年)は、cDNA-PCR法を用いて、さまざまな細胞型で組織特異的遺伝子が常に基本的な転写レベルを持つことを発見し、この現象を「非正統的転写」と名付けました。一方で、サルカーとゾンマー(1989年)はこれを「異所性転写」と呼び、非筋細胞にも筋特異的なDMD遺伝子の転写産物が存在することを示しました。この異所性転写により、筋ジストロフィー患者の診断や遺伝子欠損の解析に有用な情報が得られる可能性が示唆されました。
Kaplanら(1992年)は、異常転写が記録されている17の遺伝子をリストアップし、そのうち9つが遺伝性疾患の診断に使われていました。しかし、Robertsら(1993年)は異常転写の忠実度が低いことを指摘し、注意が必要であると報告しました。彼らは、末梢血リンパ球増多から得られたジストロフィン転写産物の50%に新規エクソンが含まれており、この新規配列は癌胎児性抗原遺伝子ファミリーの保存領域に類似しているとしました。これは、異所性転写が必ずしも組織特異的転写と同様に処理されるわけではないことを示唆しています。
● 脳におけるジストロフィン
Chamberlainら(1988年)の研究により、脳にジストロフィンmRNAが存在することが確認され、DMD患者における精神発達障害の説明に繋がる可能性が示されました。ジストロフィンの脳型転写は、筋肉型プロモーターとは異なる脳特異的プロモーターによって行われており、Chellyら(1990年)は、この脳型プロモーターが特にニューロンで非常に特異的であることを実証しました。筋肉型プロモーターは、広範な細胞で活性化されますが、脳型プロモーターは主に神経細胞で活性化されています。
● シナプス後部におけるジストロフィンの役割
脳内のジストロフィンは、シナプス後部密度に局在しており、この構造がシナプス後部の受容体を固定し、シグナル伝達を助けてシナプスの安定性を維持していると考えられています。キム氏ら(1995年)は、デュシャンヌ型筋ジストロフィー患者の脳では、この427kDのジストロフィンがシナプス後密度から消失していることを確認しました。対照群の脳では正常に存在していたため、この欠損がDMD患者の神経学的症状に寄与している可能性が示唆されています。
Barら(1990年)は、DMD遺伝子から転写される6.5kbのmRNAを同定しました。このmRNAは、脳を含む多くの非筋肉組織における主要なDMD遺伝子産物とされており、別のプロモーターによって制御されています。続いて、Boyceら(1991年)は脳におけるジストロフィンの代替5’末端をコードするゲノム領域を特定しました。この代替プロモーターは、筋肉で使われるプロモーターから90kb以上離れた位置に存在し、スプライシングされるエクソン2からも400kb離れていることがわかりました。このように物理的に大きく離れたプロモーターの存在が、脳または筋肉におけるジストロフィン発現が患者ごとに異なる可能性を示唆しています。
Lederfeinら(1992年)は、この6.5kbのmRNAの全コード配列をクローニングし、ウェスタンブロット法で対応するタンパク質を同定しました。筋肉で見られるジストロフィンとは異なり、このタンパク質は骨格筋では検出されず、非筋肉組織においてジストロフィンと同等のレベルで存在していることが確認されました。このタンパク質は、ジストロフィンのC末端およびシステインに富むドメインを持ち、選択的スプライシングによる修飾が加えられていますが、スペクトリン様反復ドメインやアクチン結合N末端ドメインが欠如しています。
● Dp71とその役割
Dp71は、DMD遺伝子の主要な非筋肉産物であり、特に成体の脳で多く発現します。Sarigら(1999年)は、Dp71の役割を調べるために、ノックアウトマウスを用いてその発現を研究しました。結果として、神経系、眼、肺、血管など、さまざまな組織でDp71のプロモーターが細胞特異的かつ段階的に活性化されることが示されました。このプロモーターは、形態形成や最終分化のプロセスに関連しており、発生過程において重要な役割を果たしていると考えられます。
● アポジストロフィンとその異なる転写産物
Blakeら(1992年)は、DMD遺伝子座から発現する4.8kbの転写産物を発見しました。この転写産物は、シュワン細胞で特に豊富に発現しており、ジストロフィンとは異なる発現パターンを示します。Blakeらはこの転写産物をアポジストロフィン-1と名付けました。その後、Tinsleyら(1993年)は、アポジストロフィン-2およびアポジストロフィン-3と呼ばれる別の転写産物を同定し、アポジストロフィンが異なる組織や発生過程において多様な機能を持つ可能性を示しました。
AhnとKunkel(1993年)は、DMD遺伝子の発現が精巧な転写およびスプライシング制御の下にあり、少なくとも5つの独立したプロモーターが異なる細胞や発生段階で転写を制御していることを指摘しました。これらのプロモーターの一部は、完全長のジストロフィンを発現し、他はC末端のドメインを発現します。また、選択的スプライシングにより、ジストロフィンに多様な機能が生じることも示唆されました。
TorelliとMuntoni(1996年)は、ジストロフィンのエクソン4が骨格筋や心筋でスプライシングアウトされても機能に問題がないことを報告しました。また、他の多くのエクソンも欠如している場合があり、それでもジストロフィンの機能が維持される可能性を示しています。この情報は、将来的にミニ遺伝子を構築し、遺伝子治療に利用できるかもしれないと述べています。
Bakkerら(1987年)とDarrasとFrancke(1987年)は、生殖細胞でのモザイク現象に関連するDMD遺伝子の欠失が複数の世代にわたって伝達されることを報告しました。このモザイク現象により、遺伝的変異が部分的に引き継がれることが示され、特定の家系でDMDの遺伝的診断が複雑化する可能性が示唆されました。
また、NormanとHarper(1989年)は、デュシャンヌ型およびベッカー型筋ジストロフィーのキャリア女性の約2.5%が症状を発症することを報告しました。このキャリア女性の有病率は、常染色体劣性肢帯型筋ジストロフィーと同程度と推定されました。さらに、女性キャリアは、ふくらはぎの肥大や筋力低下、クレアチンキナーゼの上昇がみられる場合、家族歴がなくてもDMD遺伝子のキャリアである可能性が高いと指摘しました。
Kunkelら(1985年)は、減数ハイブリダイゼーション法を使用して、DMD遺伝子の染色体欠失がホモ接合体またはヘテロ接合体の患者における特定のDNA断片をクローニングしました。彼らは、Franckeら(1985年)が報告したXp染色体の微小中間部欠失を持つ患者のDNAを解析しました。また、Bakkerら(1985年)は、DMD遺伝子座に関連する11個のRFLPマーカーを使用し、DMD保因者の診断を行いました。この技術により、妊娠12週目で99%以上の確率で男性胎児のDMD罹患が診断されました。
Rayら(1985年)は、X;21転座における転座切断点をクローニングするため、rRNA配列を使用し、DMD患者における染色体欠失を明らかにしました。続いて、BoydとBuckle(1986年)やNevinら(1986年)は、Xp21.2領域の切断点を含む複数の転座事例を報告しました。
Bodrugら(1987年)は、X;21転座を持つ症例において転座部位をクローニングし、X染色体および21番染色体から小さなDNAが失われていることを発見しました。この研究は、転座に関与する可能性のある酵素認識部位の存在を示唆しました。
チェリーら(1988年)は、DMDに関連するXp21染色体の欠失を報告し、パルスフィールドゲル電気泳動とサザンブロット法を用いて、約4メガベースの欠失を特定しました。これにより、DMD、グリセロールキナーゼ欠損症、副腎低形成に関連する遺伝子座が明らかになりました。
Feenerら(1991年)は、DMD遺伝子の「脳プロモーター」近くにある(CA)nポリモルフィズムを発見しました。この多型は、連鎖解析に有用であるとされました。
Towbinら(1991年)は、X連鎖拡張型心筋症患者の心筋でジストロフィンの発現が低下している一方、骨格筋では正常なジストロフィンが存在することを示しました。
Zhangら(2007年)は、DMD患者のリンパ球増多から分離したジストロフィンmRNAを増幅し、新たに7つの隠れエクソンを特定しました。これらのエクソンは、mRNAに保持されているため、生理学的役割がある可能性があります。
● まとめ
– 非正統的転写(異所性転写)は、筋以外の細胞でもDMD遺伝子の不完全な転写産物が見られる現象です。これにより、遺伝性疾患の診断や研究に新しい方法が提供されていますが、異常転写には注意が必要です。
– 脳におけるジストロフィンの存在は、DMD患者の知的障害との関連があり、特にシナプス後部での役割が注目されています。
– ジストロフィンの代替プロモーターによる制御は、脳や筋肉での発現に違いをもたらし、特定のプロモーターの欠失が筋ジストロフィーやX連鎖精神発達障害の原因になる可能性があります。
– Dp71は、DMD遺伝子の非筋肉組織での主要産物であり、発生過程や形態形成において重要な役割を果たしています。
– アポジストロフィンは、ジストロフィンの関連タンパク質として、異なる発現パターンを示し、組織特異的な機能を持つことが示唆されています。
– DMD遺伝子は、複数のプロモーターと選択的スプライシングにより、多様な機能を持つ転写産物を生成します。
– 生殖細胞モザイクにより、DMDの遺伝的伝達が複雑化する可能性があり、家族歴がない場合でも注意が必要です。
– キャリア女性が症状を発症することがあり、その頻度は比較的低いものの、診断には慎重な評価が求められます。
– DMD遺伝子の染色体欠失は、転座や欠失に関連して特定のDNA領域が失われることが多く、これらの欠失は遺伝子診断や病因解明に重要な情報を提供します。
– 隠れエクソンや多型の発見は、DMD遺伝子の転写調節やその複雑な機能に関する新たな知見をもたらしました。
– 心筋と骨格筋でのジストロフィン発現の違いは、特定の筋ジストロフィーの病態に関連する可能性があります。
進化
1. 西アフリカにおいて、ラッサウイルス感染に関連するLARGE(603590)とDMD(デュシャンヌ型筋ジストロフィー遺伝子)が自然選択の影響を受けたとされています。
2. ヨーロッパでは、皮膚の色素形成に関わるSLC24A5(609802)とSLC45A2(606202)が、選択を受け、これらは皮膚の色素沈着に関与しています。
3. アジアでは、毛包の発達に関わるEDAR(604095)とEDA2R(300276)が、同じく選択を受けた遺伝子として見つかっています。
これらの遺伝子は、それぞれの地域での環境適応や感染防御などの自然選択圧に関連しており、特定の生物学的プロセスにおける進化的適応が示唆されています。
遺伝子の機能
研究では、一過性の燐光異方性を使用して、両方のタンパク質がアクチンフィラメントの曲げやねじれに及ぼす影響を調べました。その結果、ジストロフィンとユトロフィンの両者がアクチンの動きを制限し、その速度を増加させることが示されました。しかし、特にアクチンが飽和状態にある場合、ユトロフィンはジストロフィンよりもアクチンのねじれ剛性を大きく減少させる効果がありました。一方で、どちらのタンパク質もアクチンの凝集や束状化には影響を与えませんでした。
このことから、Prochniewicz氏らは、ジストロフィンとユトロフィンが単にアクチンの重合を阻害するだけでなく、アクチンフィラメントが伸展やねじれにさらされても、それに対する切断抵抗を提供する可能性があるという仮説を提唱しました。
分子遺伝学
続いて、Koenigら(1987年)は、DMD遺伝子が少なくとも60個のエクソンから構成されることを発見しました。特に、この遺伝子の大部分の欠失は、わずか2kbに集中していることが分かりました。また、Worton(1987年)は、DMD遺伝子での欠失に加え、重複も発見しました。これらは、遺伝子内での不等交差によって生じた可能性が指摘されています。
さらに、Tennysonら(1995年)は、DMD遺伝子が2,300kb(2.3Mb)にわたる79個のエクソンで構成されていることを示しました。彼らは定量的RT-PCRを用いて、完全な転写が約16時間かかることを推定し、転写が進行しながらスプライシングが同時に行われるという転写共役スプライシングの存在を確認しました。
Hartら(1987年)は、ベッカー型筋ジストロフィー(BMD)の患者33人を調査し、3人にDXS164の欠失が見られ、DMDにおける欠失の頻度と同程度であることが示唆されました。
MonacoとKunkel(1987年)は、DMD/BMD遺伝子座に関する総説を行い、欠失率は他のX連鎖疾患(レッシュ・ナイハン症候群、オルニチントランスカルバミラーゼ欠損症、血友病A)に類似していることを報告しました。また、DMD遺伝子の転写方向が短腕のセントロメアから末端に向かうことが予測されました。
これらの研究は、DMD遺伝子が非常に大きな構造を持ち、エクソンとイントロンの配置が複雑であること、さらには不等交差によって遺伝子の欠失や重複が引き起こされる可能性が高いことを示しています。
Tuffery-Giraudら(2009年)は、フランスにおけるDMD遺伝子の変異に関するデータベースの報告を行い、2,046人の男性患者と38人の女性患者から得られた2,084件の独立した変異を含む2,411件のエントリーがまとめられました。このデータは、フランスでの「ジストロフィン症」の遺伝子診断の頻度が100万人中39件であることを示しています。データベース内の変異には、1,404件の大規模欠失、215件の大規模重複、465件の小規模再配列が含まれており、約40%がナンセンス変異でした。また、全体の約24%が新規変異であることが確認されています。ベッカー型筋ジストロフィー(BMD)の頻度はデュシャンヌ型筋ジストロフィー(DMD)の約43%であることも分かりました。
Oshimaら(2009年)の研究では、DMD変異のインデックス症例624例中、238例(38.1%)でゲノム再編成が確認されました。欠失は188例(79.0%)で、特にエクソン45からエクソン52間、およびエクソン8からエクソン13間に集中していました。重複は44例(18.5%)で確認され、複雑な再配列は6例(2.5%)で検出されました。さらに、再配列のメカニズムとして、非相同末端結合(NHEJ)やテンプレートスイッチング(FoSTeS)などが関与している可能性が指摘されています。
Takeshimaら(2010年)の研究では、日本のDMD/BMDデータベースに登録された442人の患者全員にDMD遺伝子の変異が確認されました。270人(61%)で欠失、38人(9%)で重複、69人(16%)でナンセンス変異、24人(5%)でスプライス部位変異が見られました。また、34人(8%)に小規模な欠失や挿入変異が認められました。日本のデータに基づき、エクソン・スキップ治療がジストロフィン症の治療における最優先事項とされました。
これらの研究により、DMD遺伝子の変異がさまざまなタイプや位置で発生し、その治療に向けたアプローチが進められていることが明らかになっています。
欠失
DMD(デュシャンヌ型筋ジストロフィー)遺伝子における最も一般的な変異は、1つ以上のエクソンの欠失であり、患者の約60~65%に見られることが報告されています【Oshima et al., 2009】。これはDMDやベッカー型筋ジストロフィー(BMD)患者に共通して見られる特徴で、ジストロフィン遺伝子の広範な部分が欠失することで引き起こされる疾患です。
Gospeら(1989年)は、ジストロフィン遺伝子の欠失が原因とされる非進行性筋疾患の家族について報告しました。この家族の男性9人は、クレアチンキナーゼ(CK)レベルの上昇や筋肉の肥大が見られたものの、筋力低下はなく、症状は進行しないことが特徴でした。DNA解析では、ジストロフィン遺伝子の最初の3分の1に欠失がありましたが、ウェスタンブロットではジストロフィンの量は減少しておらず、正常よりも小さいタンパク質が検出されました。
Doriguzziら(1993年)は、9歳で筋痛とミオグロビン尿症を示した患者について報告し、この患者のゲノムDNA解析により、エクソン45から48の欠失が確認されました。
さらに、MinettiとBonilla(1992年)は、ジストロフィンの欠損と正常な線維がモザイク状に混在するパターンを観察し、これがDMD保因者だけでなく、BMD保因者や家族性X連鎖ミオパチーの保因者にも共通する所見であることを示しました。
遺伝子解析技術の進展により、DMDやBMDに関連する変異がより詳細に研究されるようになりました。例えば、Lindlofら(1988年)の研究では、ベッカー型筋ジストロフィー(BMD)家系の36%に欠失が見られ、デュシャンヌ型筋ジストロフィー(DMD)家系では8%に欠失が確認されました。
また、Readら(1988年)は、DMDおよびBMD患者の60%に1つ以上のエクソンの欠失があることを明らかにしました。BMD患者の多くは、エクソンの欠失が小規模で、同じグループのエクソン欠失がDMDを引き起こしていることが分かっています。
これらの研究は、DMDおよびBMDの原因となる欠失の位置や規模がさまざまであり、同じ欠失でも患者の症状や進行度が異なることが多いことを示しています。また、欠失に加えて、ジストロフィン遺伝子の重複や再編成もこれらの疾患の原因となることが明らかにされています。
デュシャンヌ型筋ジストロフィー(DMD)の原因となるジストロフィン遺伝子における最も一般的な変異は、1つ以上のエクソンの欠失であり、患者の約65%に認められます。この欠失は、遺伝子内の2つのホットスポット(変異が起こりやすい領域)に集中しています。1つは近位部分に位置し、欠失の約30%がここで発生します。もう1つは遠位部分に位置し、約70%の欠失が発生しています。
● 欠失の発生頻度と遺伝パターン
研究によれば、近位部分の欠失は主に家族性のDMD症例に多く見られ、遠位部分の欠失は孤立性の症例に多いとされています。Passos-Buenoら(1992年)の研究では、近位欠失が胚発生の初期に起こりやすく、そのため家族性の症例となりやすい一方、遠位欠失は発生の後期に起こるため孤立性の症例となる可能性が高いとしています。また、近位欠失による新規突然変異には約30%の再発リスクがある一方、遠位欠失では約4%の再発リスクがあるとされています。
● 欠失の発生メカニズム
DMD遺伝子内の欠失は、特にイントロン44などの大きなイントロンに関連していることが多く、イントロン44はDMDにおける欠失の約30%でブレークポイントとなっています(Baumbachら、1989年)。また、Pizzutiら(1992年)は、第43イントロン内で発生した欠失がTHE-1ファミリーに属するトランスポゾン様要素に関連していることを発見しました。この要素は、ヒトのレトロトランスポゾンに属し、ゲノム中に1万回も存在するため、欠失の引き金となる可能性があります。
● 特異な転座事例と欠失
DMDに関連する転座も報告されています。GiacaloneとFrancke(1992年)は、X染色体と4番染色体の相互転座が原因でDMDを発症した患者について研究し、この転座がDMD遺伝子内の18kbのイントロン16を破壊していることを発見しました。この転座により、父親由来のX染色体にあった遺伝子が欠失し、結果的に発症に至ったとされています。
● 遺伝的モザイクと欠失
一部のDMD患者では、遺伝的モザイクの影響が観察されることがあります。Hoopら(1994年)は、DMDを患う2人の男性従兄弟が異なる欠失変異を有していることを発見しました。1人の従兄弟では、近位と遠位の2つの非連続欠失が確認されましたが、もう1人の従兄弟では近位欠失のみが認められました。この違いは、母親のジストロフィン遺伝子内で起こった組み換えにより、近位の欠失が「修復」され、息子に伝わったためと説明されています。
● 欠失の影響と臨床的特徴
一部の患者では、欠失の範囲や場所により、筋ジストロフィーの進行具合が異なります。Takeshimaら(1994年)は、日本人少年において、ジストロフィン遺伝子のエクソン3からエクソン41にわたる大きな欠失が確認され、これが筋ジストロフィーの中間的な症状を引き起こしていると報告しました。欠失により、ジストロフィンのアクチン結合ドメインが欠如し、重症化に寄与している可能性が示唆されています。
また、Muntoniら(1993年)は、DMD遺伝子のプロモーター領域および第1エクソンの欠失がX連鎖拡張型心筋症(CMD3B)を引き起こすことを証明しました。
重複
デュシャンヌ型筋ジストロフィー(DMD)およびベッカー型筋ジストロフィー(BMD)におけるジストロフィン遺伝子の部分的な重複は、疾患の原因となる変異の約6〜7%を占めています。これらの重複は、主にエクソンを含む領域で発生し、遺伝子の正常な構造や機能を妨げることで筋ジストロフィーを引き起こします。
● 重複の発見と遺伝メカニズム
Huら(1988年、1989年)は、DMDやBMD患者のジストロフィン遺伝子においてエクソンの重複があることを報告し、これが疾患の原因であることを証明しました。彼らは、発端者とその母親、場合によっては姉妹にまで重複が伝わっていることを確認し、これらの重複が祖父由来の不等姉妹染色分体交換によって生じた可能性を示しました。
また、HuとWorton(1992年)は、筋ジストロフィーだけでなく、他の遺伝疾患にも部分的な遺伝子重複が関連していると報告しています。遺伝子重複は、エクソンのリーディングフレームが維持されている場合はベッカー型筋ジストロフィー(軽度の表現型)として現れ、フレームシフトが起こるとデュシャンヌ型筋ジストロフィー(より重度の表現型)を引き起こす傾向があります。
● 重複の特徴と影響
ベッカー型筋ジストロフィー患者では、アンジェリーニら(1990年)が40万塩基対以上に及ぶDMD遺伝子の重複を発見し、この重複によりジストロフィンタンパク質が通常よりも大きな約600kDのサイズで存在することを示しました。これは、ジストロフィン遺伝子が100以上のエクソンを持つ長いmRNAをコードしているためです。このジストロフィンは「フリウリジストロフィン」と呼ばれ、詳細に研究されました。
● 遺伝子重複のメカニズム
ジストロフィン遺伝子の重複は、一般に不等姉妹染色分体交換によって生じると考えられていますが、一部の重複は非相同末端結合によるものであることが示唆されています。Whiteら(2006年)の研究では、エクソン2の重複が最も多く見られ、この重複の発生機構が合成依存性の非相同末端結合である可能性が示されました。
● 臨床的および遺伝的意義
重複変異の頻度は、遺伝子の5’末端付近で高く、特にエクソン2に集中しています。重複がジストロフィン遺伝子のフレームシフトを引き起こす場合、重度の症状が現れる可能性が高く、非フレームシフトの場合は軽度の症状となります。このため、重複の検出は、DMDやBMDの診断や家族内発症リスクの予測において重要です。
点突然変異
Lenkら(1993年)は、デュシャンヌ型およびベッカー型筋ジストロフィー(DMD/BMD)患者26人を対象に、ジストロフィン遺伝子のエクソン60-79をPCR-SSCP分析で調べ、7つの点突然変異と1つのイントロン多型を特定しました。DMD患者で発見された6つの点突然変異は、早期の翻訳終結を引き起こすと予測され、一方で、BMD患者で見つかった点突然変異はアミノ酸の置換を引き起こしました。特に、DMD患者の5人で精神発達障害が確認され、翻訳読み取り枠の崩壊が関与している可能性が示唆されました。この研究では、ジストロフィンのC末端に由来するタンパク質であるDp71やDp116が、DMD患者の精神発達障害に関連している可能性が指摘されています。
Lederfeinら(1993年)は、Dp71が多くの細胞や組織で発現していることを示し、プロモーターの特徴を明らかにしました。Dp71の5′-非翻訳領域は単一のエクソンから構成されており、プロモーターにはTATAボックスがないものの、GC含量が非常に高く、複数のSp1結合部位が含まれていました。このプロモーターは、ジストロフィンの筋肉型および脳型プロモーターとは物理的に離れているため、ほとんどのDMD患者においてDp71の発現には影響がないと結論付けました。
Robertsら(1992年)は、DMD患者の3分の1で見られる大規模な遺伝子再編成がないケースについて、ジストロフィンmRNAのネステッド増幅と化学的不一致検出を用いた解析を行い、7人の患者全員に早期終止コドンが見られる点突然変異が確認されました。これにより、ジストロフィンタンパク質の欠損を引き起こす可能性が示唆されました。
また、Kilimannら(1992年)は、DMD患者60人を対象に点突然変異を探索し、2つのフレームシフト変異と4つの多型を発見しました。これらの変異は、いずれもジストロフィンタンパク質の欠損を引き起こすものでした。
さらに、Robertsら(1994年)は、ジストロフィン遺伝子における小規模な変異の解析により、DMDの点突然変異の大部分がナンセンス変異であり、これが早期の翻訳終結を引き起こしていることを確認しました。
ジストロフィン遺伝子の欠失および点突然変異は、翻訳読み取り枠が維持されるかどうかによって、DMDまたはBMDを引き起こします。De Angelisら(2002年)は、エクソン51のスプライシングを制御することで、DMD患者の一部でジストロフィン合成が回復する可能性を示し、部分的な表現型の改善を達成しました。
Aartsma-Rusら(2004年)は、異なるエクソンのスキップを誘導するアンチセンスオリゴヌクレオチド(AON)の開発が、DMD患者の75%以上に有益であると報告しました。
Van Essenら(2003年)は、デュシャンヌ型筋ジストロフィー(DMD)男性患者において、母親がモザイクであることが確認されたDMD遺伝子の点突然変異を特定しました。彼らは、DMDにおける生殖細胞系モザイクによる点突然変異の報告例が過去にわずか2例しかないことを指摘しています。このようなモザイク現象は、DMDの遺伝形式において重要な役割を果たす可能性が示唆されています。
Buzinら(2005年)は、DMD患者141人を対象に、標準的な多重PCR検査では大きな欠失が確認されなかったケースに対して包括的な突然変異スキャンを実施しました。このスキャンでは、すべてのコードエクソン、イントロンのスプライス部位、およびプロモーター配列を解析し、患者の90%において原因となる変異を特定しました。そのうち、70%がタンパク質の短縮を引き起こす点変異、13%が重複、7%が欠失でした。彼らは、点変異に関連する突然変異率や、特にCpGジヌクレオチドでの変異の多さを強調し、DMD遺伝子における複雑な挿入・欠失(indel)が一般的な変異タイプであると結論づけました。
Milasinら(1996年)は、X連鎖性拡張型心筋症(XL-CMD)の家族におけるジストロフィン遺伝子のスプライス部位での点突然変異を特定しました。この変異は心筋症の原因として知られ、スプライシングの異常がDMDだけでなく心筋症にも関連していることを示唆しています。
また、Yagiら(2003年)は、無症候性ジストロフィン症候群の少年において、DMD遺伝子のイントロン2に点突然変異を発見しました。この変異は、ジストロフィンmRNAに新たなエクソンを組み込むスプライシングアクセプター部位を生成し、ヒト疾患における新たな分子メカニズムであると述べています。
Nishiyamaら(2008年)は、DMD遺伝子におけるナンセンス変異が部分的なエクソンスキップと選択的スプライシングを引き起こし、軽度の表現型に影響を与えることを確認しました。同じナンセンス変異を持つ患者でもスプライシングの違いが見られ、個人差が存在することが示唆されています。
Legardinierら(2009年)は、DMDタンパク質のロッドドメインリピート23(R23)領域における2つの変異がジストロフィンの安定性や再折りたたみに与える影響を分析し、変異によるジストロフィンの構造的および機械的特性の喪失が筋細胞の欠陥につながる可能性を示しました。
Habaraら(2009年)は、DMD遺伝子のスプライス部位における変異がエクソンスキップや隠れたスプライス部位の活性化を引き起こすことを確認し、その影響を予測する要因として、アクセプタースプライス部位の強度とエクソンの長さが重要であると結論づけました。
遺伝子型と表現型の相関
Forrestら(1987年)は、欠失検出率を40%にまで引き上げ、欠失が特定の領域で頻発していることを示しました。このことは、DMDやBMDにおけるエクソン欠失の優先性を示唆しています。さらに、欠失を有する家族では異なる表現型が見られ、ジストロフィン欠失に対する個人差があることが示されました。
Hoffmanら(1987年)は、DMDではジストロフィンが完全に欠如しているのに対し、BMDでは異常な短縮型ジストロフィンが合成されることを証明しました。Monacoら(1988年)は、DMDではフレームシフトが起こりジストロフィンが合成されないのに対し、BMDではリーディングフレームが保たれ、部分的に機能するジストロフィンが合成されることを示しました。
Koenigら(1989年)は、DMD/BMDにおける「リーディングフレーム」理論の妥当性を258の欠失例で確認し、92%の症例で表現型との一致を見出しました。この理論は、DMDとBMDの診断や予後判定において重要な意義を持ちます。
Daviesら(1988年)は、表現型の重症度と欠失の大きさは相関しないと結論づけ、DMDとBMDの表現型差異はフレームシフトの有無に起因することを示しました。
Dubrovskyら(1995年)は、DMDと筋強直性ジストロフィーを併発した患者を報告し、ジストロフィン逆転線維や体細胞モザイクが症状の進行に影響を与える可能性を示唆しました。
これらの研究により、DMDおよびBMDにおけるジストロフィン欠失と表現型の関連が明確化され、遺伝診断の精度が向上しました。
ジストロフィン遺伝子は、少なくとも5つの異なるプロモーターを持ち、組織特異的にアイソフォームの転写を制御しています。3つのプロモーターは遺伝子の5’末端に位置し、427kDの脳および筋肉アイソフォームを生産します。さらに、2つのプロモーターは遠位部分に存在し、代替的な第1エクソンから始まるDp116およびDp71という分子量の異なるアイソフォームを生成します。特に、Dp116は末梢神経のシュワン細胞に発現し、Comiら(1995年)は、ジストロフィンとDp116の欠損が神経筋疾患と関連していることを報告しました。ある患者では、精神発達障害、筋力低下、脳の異常が認められ、Dp116の欠損が確認されました。
TodorovaとDanieli(1997年)は、BMD遺伝子における72の単一塩基変異を調査し、CpG変異の頻度は他の遺伝子と比較して低い一方、トランジション(塩基の変換)は予想よりも高頻度であることを発見しました。彼らは、特定の配列モチーフが変異発生に関与している可能性を示唆し、直鎖反復や逆鎖反復が重要な役割を果たしていると結論づけました。
X連鎖性拡張型心筋症(CMD3B)では、吉田ら(1993年)がDMD遺伝子のエクソン1にヒトL1要素が挿入されたユニークな例を報告しました。この挿入により、ジストロフィンの転写や安定性に影響が生じ、心筋症が発症しました。患者は心電図異常と高CK血症を呈し、拡張型心筋症に進行しましたが、骨格筋の症状は認められませんでした。
さらに、Howardら(1998年)は、ジストロフィンのアイソフォームであるDp260とDp71が網膜の異なる部分に局在しており、βジストログリカンと相互作用していることを明らかにしました。これらのアイソフォームは、網膜の電気生理学にそれぞれ異なる役割を果たしています。
ヒトにおいても、Pillersら(1999年)は、DMD/BMD患者での遺伝子変異の位置と網膜電図(ERG)異常の相関を調査しました。Dp260の変異は、網膜の電気生理学に影響を与えることが確認されました。
Moizardら(2000年)は、精神発達障害を持つDMD患者におけるDp71転写体の変異を分析し、早期翻訳終結を引き起こす点突然変異が重度の精神発達障害と関連していることを発見しました。
これらの研究は、ジストロフィン遺伝子が多様なプロモーターを通じて組織特異的に異なるアイソフォームを生み出し、それが筋疾患や心疾患、精神発達障害に寄与することを示しています。
Ginjaar ら(2000年)は、X連鎖筋ジストロフィーの3人の男性が異なる表現型を示す家族について研究し、その違いはDMD遺伝子のエクソン29のスキップのレベルに関連している可能性を示唆しました。これは、異なるスプライシングの効率が病気の重症度に影響を与えることを示しています。
Greener ら(2002年)は、DMD遺伝子の3’UTR(未翻訳領域)が、進化的に高度に保存されていることを示しました。彼らは、両生類やサメなどの動物から対応する配列を特定し、BMD患者において、3′-UTRに関連する変異が影響を及ぼす可能性を示唆しています。特に、BMD患者のエクソン79および3′-UTRの欠失が報告されていますが、3′-UTR領域の変異はスクリーニングの対象外であることが多いです。
Tuffery-Giraudら(2005年)は、BMD患者5人のDMD遺伝子に5つのスプライス部位変異を特定しました。エクソン・スキップによるインフレーム欠失は、軽度の表現型を引き起こし、イントロン変異は複雑なスプライシングの変化を引き起こしましたが、いくらか正常な転写産物も残存していました。これらの研究は、エクソン・スキップの治療戦略に役立つ可能性があります。
Gurvichら(2008年)は、DMD遺伝子のスプライス部位変異により「偽エクソン」が形成されることがあると指摘しています。DMD患者とBMD患者では偽エクソンを含む転写物の量が異なり、この違いが病気の重症度の違いを説明できる可能性があります。また、偽エクソンに対するアンチセンスオリゴヌクレオチドを使用することで、完全長のジストロフィンの発現が回復する可能性が示されています。
Daoudら(2009年)は、DMDおよびBMD患者における精神発達障害の分子基盤を調査し、特にジストロフィンのアイソフォームであるDp71の欠損が精神発達障害と関連していることを示しました。Dp71の欠損は、IQを約2標準偏差低下させる可能性があるとされています。
Carsanaら(2005年)は、BMDおよびDMD患者108人の遺伝子型/表現型の相関性を評価し、ジストロフィン遺伝子のヒンジIII領域の欠失が、より軽度のBMD表現型と相関することを発見しました。
Wangら(2018年)は、特定のDMD遺伝子変異がエクソン・スキップの基礎レベルを高め、歩行不能になる年齢を遅らせることを発見しました。エクソン44スキップやエクソン8スキップが可能な変異を持つ患者は、他の変異を持つ患者よりも進行が遅いことが示されています。
Zambonら(2022年)は、エクソン2の重複を持つDMD患者が他の患者よりも進行が遅いことを報告し、特に歩行不能になる年齢や肺機能の低下が遅れることが示唆されています。
これらの研究は、DMD/BMD患者におけるジストロフィン遺伝子の変異がどのように病気の重症度に影響を与えるか、また治療戦略としてエクソン・スキップが有効である可能性を示唆しています。
動物モデル
Dixon ら(1988年)は、マウス胚の筋肉で異なるジストロフィンmRNAアイソフォームを発見し、これが転写開始部位やRNAプロセシングの違いによるものだと示唆しました。続くLee ら(1991年)の研究では、マウスのジストロフィンmRNA全長cDNAを合成し、ジストロフィンが細胞膜の適切な位置に局在することを確認しました。これにより、将来的な遺伝子治療の可能性が示されました。
Bittner ら(1994年)は、mdxマウスへの正常な筋肉の移植によってジストロフィンが再び発現することを確認しましたが、その後、ジストロフィンに対する抗体が生成されたにもかかわらず、移植された筋肉は拒絶されなかったことを発見しました。この結果は、他の疾患における遺伝子治療にも適用できる可能性があります。
一方、McArdle ら(1994年)は、ジストロフィンの欠如が筋形質膜の透過性を増加させるという「細胞膜透過性」仮説を否定しました。mdxマウスでは、壊死線維では透過性が増加しましたが、壊死前の線維ではその証拠は見つかりませんでした。
Cox ら(1993年)の研究では、mdxマウスに完全長のジストロフィン遺伝子を導入することで、筋ジストロフィーの異常が修正され、有害な副作用を伴わずにジストロフィンを過剰発現できることが示されました。これにより、ジストロフィー性筋疾患に対する遺伝子治療の可能性が示されました。
犬モデルの研究では、Sharp ら(1992年)が、ゴールデンレトリバー犬のX連鎖筋ジストロフィー(GRMD)におけるジストロフィン欠損の原因が、第6イントロンのスプライス部位の変異であることを示しました。これにより、スプライス異常が筋ジストロフィーの原因となることが確認されました。
さらに、Vieira ら(2015年)は、GRMD犬でJag1遺伝子の変異が筋衛星細胞の増殖能力を高め、筋ジストロフィーの症状を軽減する「エスケープ」犬を発見しました。Jag1の役割は、筋再生や治療の新たなターゲットとして注目されました。
、Tinsley ら(1996年)は、mdxマウスにおけるユトロフィンの過剰発現が筋ジストロフィーの症状を軽減することを示し、ユトロフィンがDMD治療の有望なターゲットであることを示しました。この研究は、ジストロフィンの代替としてのユトロフィンの発現増加が治療に役立つ可能性を示しています。
これらの研究は、DMDの治療戦略として、ジストロフィン遺伝子の遺伝子治療、ジストロフィンと密接に関連するユトロフィンの発現増加、スプライス異常の修正が有望であることを示唆しています。
Barton-Davisら(1999年)は、筋ジストロフィーの原因となるジストロフィン遺伝子の早期終止コドン変異に対し、アミノグリコシド系抗生物質ゲンタマイシンが有効である可能性を報告しました。mdxマウスの筋細胞にゲンタマイシンを投与すると、ジストロフィンの発現と細胞膜への局在が観察されました。これにより、ゲンタマイシンが終止コドンを抑制し、機能的なジストロフィンを再び生成する可能性が示され、最大15%のDMD患者に対して治療効果が期待されるとしました。
MankinとLiebman(1999年)は、アミノグリコシドによる治療の問題点を指摘しました。終止コドンが抑制された結果、変異したアミノ酸が含まれたタンパク質が生成され、その活性や安定性、さらに治療の長期使用による副作用が課題になる可能性があると述べました。
一方、Gussoni ら(1999年)は、mdxマウスにおける骨髄移植で、正常な造血幹細胞や筋肉由来の幹細胞が移植された筋肉にジストロフィンの発現を部分的に回復させることを確認しました。この結果は、全身的な筋肉修復の可能性を示し、DMDの新しい治療アプローチとして注目されました。
さらに、Wehling ら(2001年)は、ジストロフィン欠損による筋肉での一酸化窒素合成酵素(NOS1)の減少が炎症を悪化させると提唱し、NOS遺伝子導入によりmdxマウスの筋損傷が軽減されることを示しました。
Crawford ら(2001年)は、mdxマウスでリバータント線維と呼ばれるジストロフィン陽性細胞が存在し、これが代替スプライシングや二次突然変異によって生じることを示唆しました。この線維はジストロフィンの機能を回復する一助になる可能性がありますが、ポジティブセレクションによって維持される必要があることも明らかにされました。
遺伝子治療の視点からは、坂本氏ら(2002年)が開発したロッド切断型マイクロジストロフィンが、mdxマウスの筋病理を改善し、特に横隔膜の収縮力を正常に回復させる効果が確認されました。このマイクロジストロフィンは、アデノ随伴ウイルスベクターを使用した遺伝子治療に適していることが示唆されました。
これらの研究は、DMD治療における薬理学的および遺伝子治療の可能性を示し、特に終止コドン抑制や幹細胞移植が新しい治療戦略として注目されています。
Harperら(2002年)は、ジストロフィンタンパク質の構造的機能を詳細に分析し、ジストロフィンの特定の領域を削除したミニジストロフィンやマイクロジストロフィンが、DMDのトランスジェニックmdxマウスにおけるジストロフィーの進行を抑制することを示しました。最小のジストロフィンを発現した筋肉は、活動による損傷から完全に保護され、形態的にも正常な筋肉と区別がつかないほどでした。また、これらの短縮型ジストロフィンを運ぶアデノ随伴ウイルス(AAV)をmdxマウスに投与した結果、筋肉の病理学的特徴が劇的に改善されました。この結果は、マイクロジストロフィンを用いた遺伝子治療が、DMDに対する有効な治療手段となり得ることを示唆しています。
Xiongら(2002年)は、ジストロフィン欠損症がエンテロウイルス感染による心筋症の発症を助長するという仮説を立てました。ジストロフィン欠損マウスは、エンテロウイルス感染により重度の心筋症を発症し、ウイルスの複製や細胞からの放出が増加しました。ジストロフィンの切断抵抗性変異体を発現させた結果、ウイルスの複製と放出が抑制され、心筋症の進行が緩和されることが示されました。これにより、ウイルス感染がジストロフィン欠損による心筋症の重症度に影響を与える可能性が示唆されました。
Porterら(2002年)は、mdxマウスの筋肉における慢性炎症反応をDNAマイクロアレイ技術で解析し、多数の炎症関連遺伝子が活性化されていることを確認しました。特に、分泌性リン酸化タンパク質1(SPP1)の発現が著しく増加しており、DMD患者に見られる広範な線維症の原因が、転写後のコラーゲン制御にある可能性を示しました。
Fabbら(2002年)は、CMVプロモーター駆動のマイクロジストロフィンcDNAを含むAAVベクターを設計し、mdxマウスに導入することで、50%以上の筋線維にジストロフィンが発現し、筋ジストロフィーの症状が大幅に軽減されることを示しました。
Bogdanovichら(2002年)は、ミオスタチン阻害によるmdxマウスでの筋肉量の増加と筋損傷の減少を報告し、この方法が従来の遺伝子治療の課題を克服できる可能性を示しました。
これらの研究は、ジストロフィンの構造機能解析や遺伝子治療、さらにはミオスタチン阻害を通じたDMDの新しい治療戦略に対する重要な知見を提供しており、ジストロフィーの進行を防ぐための多様なアプローチを示しています。
Dallozら(2003年)は、網膜神経伝達における異常がジストロフィンの短い産物に関連する可能性があると仮定し、特にC末端ジストロフィン産物であるDp71の役割を調査しました。Dp71を欠損したマウスを使った研究で、網膜の電気生理学的な異常は観察されなかったものの、内境界膜(ILM)や血管周辺におけるユトロフィンの増加や、βジストログリカンの減少が見られました。また、Dp71欠損により、内向き整流カリウムチャネルKir4.1やアクアポリン-4の局在が不安定化することが明らかになり、これらのタンパク質の凝集・安定化にDp71が重要である可能性が示唆されました。
Porterら(2003, 2004年)は、mdxマウスを用いて、筋肉における時間経過に伴う遺伝子発現パターンを解析しました。ジストロフィン欠損自体は疾患の発症に必要であるが十分条件ではないことが示され、下肢筋肉では炎症や線維化、筋再生不全などの特徴が見られる一方、外眼筋ではほとんど変化がなく、筋群ごとに異なる遺伝子発現が影響を与えることが確認されました。この結果は、DMDに対する異なる筋肉の反応の根底に筋群特異的なメカニズムが存在することを示唆しています。
Moghadaszadehら(2003年)は、ADAM12を過剰発現するトランスジェニックマウスの筋再生が促進され、筋ジストロフィーの病理が軽減されることを発見しました。この研究では、α-7Bインテグリンやウトロフィンの発現が増加し、ジストロフィン関連糖タンパク質の発現も増加していたことが示され、筋再生におけるADAM12の重要な役割が示唆されました。
東岡ら(2004年)は、ジストロフィンが心筋細胞膜の安定性に関与しており、心筋ジストロフィーの進行に伴ってジストロフィンが切断され、筋細胞質へ移行することを実証しました。この現象は、急性・慢性心不全や原因不明の拡張型心筋症の患者でも確認され、心筋細胞膜の不安定性が共通のメカニズムとして関与していることが示されました。
Goyenvalleら(2004年)は、修飾されたU7小核RNAに結合したアンチセンス配列を用いることで、mdxマウスにおけるジストロフィンmRNAのエクソン・スキップを持続的に誘導し、筋肉全体で機能的なジストロフィンの発現を回復させ、筋ジストロフィーの症状を改善しました。
Yueら(2004年)は、ジストロフィンの部分的な発現を持つヘテロ接合型mdxマウスを作製し、このマウスでは心機能が維持されていることを確認しました。心筋細胞の50%でジストロフィンが発現していれば、DMDの心筋症を予防できる可能性があると結論づけました。
Abmayrら(2004年)は、酸化ストレスから細胞を保護するARCタンパク質の役割を調査しましたが、mdxマウスにおけるARCの過剰発現はジストロフィー病理を軽減しないことが示され、ARCが筋線維死に大きく関与していないことが示唆されました。
これらの研究は、ジストロフィンやその関連タンパク質の役割、遺伝子発現の変化、およびDMDの治療法の開発に対する重要な知見を提供しており、遺伝子治療やアンチセンス技術など、DMDに対する新しい治療戦略の可能性を示しています。
一部のX連鎖拡張型心筋症(CMD3B; 302045)患者では、ジストロフィン遺伝子の変異にもかかわらず、骨格筋ミオパチーの症状が見られません。これは、心筋ではなく骨格筋で、ジストロフィンの脳および小脳プルキンエ(CP)アイソフォームが発現していることが原因と考えられています。De Repentignyら(2004年)は、ジストロフィンCPプロモーターがDME1によって骨格筋で活性化されることを示しましたが、心筋では活性化されませんでした。この研究は、ジストロフィンのアイソフォーム発現が、筋組織ごとの異なる病態進行に影響を与えることを示唆しています。
安田ら(2005年)の研究では、ジストロフィン欠損症心筋細胞の伸展によるカルシウム過負荷とコンプライアンスの低下が指摘されましたが、ポロキサマー-188という膜シーラントがこれらの欠陥を修正し、急性心不全を防止できることが示されました。ポロキサマー-188は、心筋症の治療の新たなアプローチとなる可能性が示唆されています。
Guoら(2006年)は、ジストロフィンとインテグリンの二重ノックアウトマウス(DKO)が重度の筋ジストロフィーと早期死亡を示すことを報告しました。これは、これらのタンパク質が筋肉の機械的安定性に不可欠であり、欠損が心不全や筋再生の不全を引き起こすことを示しています。
Bellingerら(2009年)は、mdxマウスにおいてカルシウムチャネルRyr1の異常がカルシウム漏れを引き起こし、筋力低下や筋変性に寄与していることを明らかにしました。Ryr1チャネルの機能を修正する化合物S107は、筋機能を改善し、mdxマウスの運動能力を向上させました。
Liら(2009年)は、サルコグリカンとジストロフィンの二重ノックアウトマウスがmdxマウスよりも重症の筋ジストロフィーを示し、サルコグリカンの発現レベルが筋疾患の進行に関与していることを示しました。さらに、マトリックスメタロプロテアーゼ-9(MMP9)の阻害が筋再生を促進し、mdxマウスの筋病理を改善することも示されました。
Wehling-Henricksら(2009年)は、神経型一酸化窒素合成酵素(nNOS)の欠損がmdxマウスの易疲労性に関与していることを明らかにし、nNOSが解糖代謝に関与し、ホスホフルクトキナーゼ(PFK)の活性を調節することを示しました。nNOSの機能が失われると、解糖が障害され、筋肉の疲労が増加することが確認されました。
これらの研究は、ジストロフィンおよび関連タンパク質の欠損が筋機能に与える影響を明らかにし、カルシウム代謝やエネルギー代謝の異常が筋ジストロフィーの進行に重要な役割を果たしていることを示しています。また、これらのメカニズムを標的とする治療法の可能性も示されています。
三浦ら(2009年)は、ペルオキシソーム増殖因子活性化受容体PPAR-β/δ(PPARD)のアゴニストであるGW501516が、ユトロフィンA(UTRN)のプロモーターを介してユトロフィンAのmRNAレベルを増加させ、mdxマウスの骨格筋の筋細胞膜でのユトロフィン発現を促進することを示しました。治療により、筋細胞膜の完全性が改善し、筋損傷に対する保護効果が得られたことから、PPARDアゴニストはDMDの治療に有効な戦略である可能性が示唆されました。
Long ら(2014年)は、CRISPR/Cas9を使用してmdxマウスのDmd遺伝子を修正し、筋肉の構造と機能を回復させることに成功しました。修正された細胞の優位性により、遺伝子修正効率を上回る筋表現型の回復が見られました。彼らは、将来の技術的進歩により、DMD患者の体細胞における遺伝子修正が可能になると期待しています。
MoorwoodとBarton(2014年)は、DMD患者の筋生検で小胞体ストレスと未処理タンパク質応答(UPR)を示すCASP4が検出され、mdxマウスでも同様のストレス応答が確認されました。Casp12をノックアウトしたmdxマウスでは、筋力が回復し、筋変性が減少したため、異常なUPR活性がDMDの病態に寄与している可能性が示唆されました。
Vermaら(2010年)は、血管密度を増加させたmdxマウスモデルを作成し、筋組織の改善と筋力の回復が確認されました。また、ユトロフィン欠損マウスでも血管増加により筋組織の改善が見られ、血管新生がDMDの組織学的および機能的改善に寄与する可能性を示しました。
Amoasiiら(2018年)は、DMDの犬モデルにCRISPR遺伝子編集を導入し、骨格筋や心筋でジストロフィンの発現が回復したことを報告しました。これにより、筋組織の改善が確認され、遺伝子編集アプローチがDMDの治療に臨床的に有望であることが示されました。
これらの研究は、DMDにおけるさまざまな治療アプローチ(PPARアゴニスト、CRISPR/Cas9、血管新生促進、UPRの調節など)の可能性を示しており、筋ジストロフィーの病態を改善するための新しい戦略を提供しています。
アレリックバリアント
DMD, GLU1157TER
Bulman ら (1991) は、ジストロフィンのN末端を標的とする抗体を使用して、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者における短縮型タンパク質を同定しました。C末端に対する抗体では交差反応性物質は同定されませんでした。これはジストロフィンの翻訳が早期に終結していることと一致する結果です。126kDと推定された分子量は、mRNAおよび遺伝子における変異のおおよその位置を示唆しています。患者の筋肉の cDNA からクローン化した PCR 産物のシークエンシングと、増幅した患者のゲノム DNA の直接シークエンシングにより、3714 番目の位置で G-to-T 転移(GAG から TAG)が起こり、グルタミン酸コドン 1157 がアンバー停止コドンに変化していることが分かりました。これは、ヒトの DMD における点突然変異の最初の報告例でした。
0002 ベッカー型筋ジストロフィー、非定型
プロモーター欠失
免疫ブロット法によるジストロフィンの分析で、対照群と比較して発現量が減少していることが判明したベッカー型筋ジストロフィー(BMD; 300376)患者7人のうちの1人について、Bushby ら(1991年)はDMD遺伝子のプロモーター領域から予想される断片の1つが欠失していることを発見しました。彼らはプロモーター領域内で3組のプライマーを使用してPCRを行い、続いてドットブロット法と制限酵素分析を行いました。他のプライマーセットで増幅した際に正常なサイズの断片が検出されたことから、大きな欠失は除外されました。この患者には筋疾患の家族歴はなく、初期の運動発達と運動能力は完全に正常でした。しかし、5歳からふくらはぎと太ももにけいれん性の筋肉痛が現れ、運動能力を制限するほどひどいものでした。この痛みは拘縮とは関連しておらず、子供の場合は安静にしていれば約1時間で痛みが引きましたが、35歳になると24時間痛みが続くこともありました。激しい運動をすると尿が黒くなることがありましたが、ミオグロビン尿症は一度も証明されたことはありません。また、10代半ばから、重いものを持ち上げると手や肩に痛みを感じるようになりました。痙攣の重症度と持続時間は、Gospeら(1989年)が報告した家族の症状と類似していました。筋力の低下は、30歳頃に階段の昇り降りに困難を感じるようになって初めて気づきました。この筋力低下は徐々に進行し、40歳時には軽度の脊柱前弯歩行となり、階段を上るには手すり2本が必要となり、椅子や床から立ち上がるには太ももで体を支える必要がありました。 血清CPK値は非常に高く、心電図では不完全右脚ブロックとIIIおよびaVR誘導におけるQ波が認められました。ジストロフィンは依然として正常値の73%で産生されており、プロモーターがその機能を低下させながらも保持しているか、あるいは筋肉におけるジストロフィンの発現を制御する追加の配列が存在していることを示唆しています。 Boyceら(1991年)は、ジストロフィン筋肉プロモーターの特定の欠失によりベッカー型筋ジストロフィーを発症した患者も特定しています。
0003 デュシャンヌ型筋ジストロフィー
DMD, GLU931TER
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Roberts ら(1992年)はDMD遺伝子のヌクレオチド2999のG-to-Tトランスバージョンを特定し、グルタミン-931が「ストップ」に変化していることを明らかにしました。
0.0004 デュシャンヌ型筋ジストロフィー
DMD、GLN1851TER
中程度のデュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Roberts ら(1992)は、DMD 遺伝子のヌクレオチド 5759 における C-to-T 転移を特定し、グルタミン-1851 が終止コドンに変換されることを明らかにしました。臨床重症度は、デュシャンヌ型筋ジストロフィーとベッカー型筋ジストロフィーの中間でした。
0.0005 デュシャンヌ型筋ジストロフィー
DMD、ARG2982TER
デュシャンヌ型筋ジストロフィー(DMD;310200)の男性において、Roberts ら(1992)は、ヌクレオチド 9152 における C-to-T 変異を特定し、アルギニン-2982 が「停止」に変換されることを明らかにしました。
0.0006 デュシャンヌ型筋ジストロフィー
DMD、IVS68、T-A、+2
デュシャンヌ型筋ジストロフィー(DMD; 310200)および精神発達障害の患者において、Roberts ら (1992) は、エクソン 68(ヌクレオチド 10016-10182)のスキップを引き起こす、イントロン 68 のドナー スプライス部位における GT から GA への変化を特定しました。この患者では、転写産物の配列の欠失によりフレームシフトが起こり、翻訳が早期に終了しました。
0.0007 デュシャンヌ型筋ジストロフィー
DMD, ARG3370TER
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Roberts ら(1992)はヌクレオチド10316におけるC-to-T転移を観察し、アルギニンコドン3370が「停止」に変換されました。
0.0008 デュシャンヌ型筋ジストロフィー
DMD, EX73-76DEL
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Roberts ら(1992)はエクソン73-76(ヌクレオチド10537-11129)の欠失をもたらす点突然変異を特定しました。ロイシン3444の後にフレームシフトが生じていました。
0.0009 デュシャンヌ型筋ジストロフィー
DMD、1-BP欠失、10662T
中程度のデュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Roberts ら(1992)はチミンヌクレオチド10662の欠失により、ロイシン3485でフレームシフトと早期終結が起こることを観察しました。Lenk ら(1993)は、デュシャンヌ型筋ジストロフィー患者においてこの突然変異を特定しました。
0.0010 デュシャンヌ型筋ジストロフィー
DMD、1-BP INS、EX12
キリマンら(1992年)は、化学的不適合切断により、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、4つのT残基の連続した領域に1塩基の挿入(T)があることを特定しました。これにより、13コドン下流の新しいリーディングフレームでストップコドンによるフレームシフトが起こります。推定される翻訳産物は、ジストロフィンのアミノ酸配列のわずか13%で終了することになり、これは機能しないことが予想されます。母親は変異のキャリアでした。 .
0011 デュシャンヌ型筋ジストロフィー
DMD、AG-T、EX48
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者で、検出可能な欠失または重複がない患者において、Kilimann ら(1992)は化学的不適合切断法を用いて、正常なAGジヌクレオチドが1つのTに置換していることを特定しました。これにより、フレームシフトが起こり、新しいリーディングフレームは、エクソン49の開始点から変異から11トリプレット下流のストップコドンによって終結しました。推定されるポリペプチドは、正常なジストロフィンの長さの65%で途切れ、3重らせん棒の最後の4分の1と、独特なシステインに富むドメインおよびC末端ドメインを失っています。したがって、おそらく機能していないと考えられます。
0.0012 デュシャンヌ型筋ジストロフィー
DMD、EX21欠失
Rininsland ら(1992年)は、逆転写および PCR 法を用いて末梢血リンパ球増多における異所性転写(非正統的またはリーキー転写とも呼ばれる)を分析し、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者におけるエクソン21の欠失を特定しました。
0013 デュシャンヌ型筋ジストロフィー
DMD、EX18DEL
追加情報として、Rininsland ら(1992)は、異所性転写産物分析により、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者のエクソン18のゲノム欠失を特定したと報告しています。
0.0014 デュシャンヌ型筋ジストロフィー
DMD, GLN2319TER
デュシャンヌ型筋ジストロフィー(DMD)の2人の兄弟(310200)において、Clemensら(1992)は、アミノ酸位置2319における黄褐色の鎖終止コドンを実証しました。この変化の原因は、ヌクレオチド7163におけるCからTへの転移でした。影響を受けた男児のゲノムDNAの研究では、大きな遺伝子再配列は認められませんでしたが、エクソン48の近位部分を含むHindIIIサザンフラグメントが欠如していたため、そのエクソンに新たなHindIII制限部位を生じさせる点変異が特定されました。
0.0015 デュシャンヌ型筋ジストロフィー
DMD、ARG768TER、C-T、NT2510
デュシャンヌ型筋ジストロフィー(DMD)患者110人(310200)において、CpGジヌクレオチドを含む5つのDMD(310200)エクソンをスクリーニングするためにヘテロ二重鎖法を使用し、検出可能な欠失または重複がないことを確認したPriorら(1993)は、エクソン19に生じた2つの異なるナンセンス変異と1つの塩基欠失を特定しました。彼らは、ジストロフィン遺伝子における小規模な変異の集積に関する最初の報告であると述べています。 エクソン19のPCR産物が異常な位置に移動している患者3人が特定されました。 各ヘテロ二重鎖の配列決定により、患者1はヌクレオチド2510におけるC-to-T転移によるナンセンス変異があり、CGA(アルギニン)コドンがTGA(ストップ)に変化していることが分かりました。患者2は、2568-2570ヌクレオチドに存在する3つのシトシンのうち1つが欠失していました。その結果、フレームシフトが起こり、ロイシン787がトリプトファンに変換され、エクソン19の末端の変異から6トリプレット下流のコドン792で停止しました(300377.0016)。患者3では、ヌクレオチド2522でG-to-Tのトランスバージョンが起こり、GAG(グルタミン酸)コドンがTAG(ストップ)に変換されました(300377.0017)。エクソン19、およびスクリーニングされた他の4つのエクソン(6、7、9、14)では、中立多型は認められませんでした。
.0016 デュシャンヌ型筋ジストロフィー
DMD、1-BP欠失、2568C
300377.0015およびPriorら(1993年)を参照のこと。
.0017 デュシャンヌ型筋ジストロフィー
DMD、GLU772TER、G-T、NT2522
300377.0015およびPriorら(1993年)を参照してください。
.0018 ベッカー型筋ジストロフィー
DMD、IVS19、A-C、+3
ベッカー型筋ジストロフィー(BMD;300376)の叔父と甥がいる家族において、DMD遺伝子の部分欠失が認められた。Laing et al. (1992)は、叔父の姉の人工妊娠中絶で流産した胎児の遺伝子に完全欠失が認められたことを発見した。マーカーにより、2つの症例で同じX染色体が影響を受けていることが示されました。Laingら(1992年)は、偶然の出現と前駆突然変異の可能性について論じました。この家族に関するその後の研究で、Wiltonら(1993年)は、ベッカー型筋ジストロフィーの原因となる主な突然変異はスプライス部位の変化であることを実証しました。この家族のBMD患者の一人の筋生検から採取したRNAをRT-PCR法で分析し、成熟遺伝子転写物を研究しました。ジストロフィンmRNAからエクソン19が欠失していましたが、ゲノムレベルでは存在していました。エクソン19の欠失は、エクソン19の5’スプライスサイトの3番目のヌクレオチドにおけるA-to-C転換と関連していることが分かりました。この患者では、正常なサイズのジストロフィンmRNAおよびジストロフィンの発現レベルが低かった。前述の通り、ゴールデンレトリバー犬の筋ジストロフィーの原因はスプライス部位の突然変異です(Sharp et al., 1992)。 .
0019 ベッカー型筋ジストロフィー
DMD、IVS57、G-C、-1
ロバーツら(1993年)は、31歳で初めて受診した際、走ることや階段の昇降が困難で、頻繁に転倒していた、ベッカー型筋ジストロフィー(BMD; 300376)の日本人系男性のDMD遺伝子における点突然変異について報告しています。診察の結果、ふくらはぎの肥大と上肢の筋力低下が認められ、ゴワーズ法で床から体を起こしていました。母方の叔父は進行性筋ジストロフィーを患い、18歳で心不全により急死しました。発端者は36歳で車椅子生活となり、43歳で心不全により死亡しました。エクソン57のスプライスアクセプター部位における1塩基置換により、コンセンサス配列AGがACに変換され、このエクソンの異常スプライシングが起こりました。転写産物の約半数はエクソン57のフレームシフトによる欠失(リピートドメイン22以降の早期翻訳終結をもたらす)を受け、残りの約半数は通常のスプライスアクセプターサイトの3塩基上流18bpの隠れたスプライスアクセプターサイトを使用しているように見えました(野生型ジストロフィンとアミノ酸6個の中間部欠失のみが異なる翻訳産物が生成されると予想されます)。
0.0020 デュシャンヌ型筋ジストロフィー
DMD、LEU54ARG
Prior ら(1993年)は、ヘテロ二重鎖技術と増幅産物の直接シークエンシングを用いて、デュシャンヌ型筋ジストロフィー(DMD; 310200)の105人の欠失/重複ではない患者を対象に点突然変異のスクリーニングを行い、そのうちの1人から、DMDの原因として検出された最初のジストロフィンのミスセンス変異であると考えられる変異を発見しました。この突然変異は、T-to-G 転換であり、アクチン結合ドメインのアミノ酸54番目の、進化上保存されているロイシンがアルギニンに置換されていました。 生成されたジストロフィンタンパク質は、適切に局在し、DMD患者で観察されるよりも高いレベルで存在していました。 このことは、タンパク質の安定性には完全なアクチン結合ドメインが必要であり、機能には不可欠であることを示唆しています。
0021 拡張型心筋症、3B
DMD、EX1DEL
4人の兄弟と2人の母方の叔父が拡張型心筋症(CMD3B;302045)であった家族において、Muntoniら(1993)はDMD遺伝子の最初の筋エキソンの欠失と筋プロモーター領域について報告しています。骨格筋の筋力低下は認められませんでしたが、血清中のクレアチンキナーゼ値は上昇していました。彼らは、「脳プロモーターが、骨格筋では比較的高い転写レベルを駆動しているが、心臓では駆動していない」という仮説を提唱しました。Bies(1994年)とTowbinおよびOrtiz-Lopez(1994年)は、筋プロモーターの欠失が心筋症に特異的であるかどうかについて疑問を呈しました。Muntoni ら(1995)は、この欠失は脳またはプルキンエ細胞のプロモーターを除去しないと述べています。欠失により筋アイソフォームの転写開始部位が除去されたにもかかわらず、Muntoni ら(1993)の家族の骨格筋では免疫細胞化学的にジストロフィンが検出されました。これらの患者の骨格筋においてジストロフィン転写を促進しているプロモーターを特定するために、Muntoni ら(1995)はまず、正常なヒトの骨格筋および心筋、ならびにマウスの脳および小脳における、筋、脳、およびプルキンエ細胞アイソフォームのエクソン1の発現を評価しました。健常者では、脳アイソフォームのわずかな発現を除いて、骨格筋では筋肉アイソフォームのみが著しく転写されていることが分かりました。一方、心筋ではエクソン1の筋肉および脳アイソフォームの両方が高度に発現しています。一方、X連鎖拡張型心筋症患者の骨格筋では、脳型およびプルキンエ細胞型の両アイソフォームが発現していました。 骨格筋におけるこれら2つのアイソフォームの過剰発現は、男性患者の筋疾患を防ぐ上で極めて重要であると考えられます。 心筋症が重症化する理由は、心臓におけるジストロフィンのデータが欠如しているため、推測の域を出ません。患者では、イントロン1の5’領域の調節配列が欠失しており、これがさまざまな組織におけるジストロフィンの発現に重要な意味を持っている可能性があります。また、この家族における欠失は、2つのジストロフィンアクチン結合ドメインのうちの1つだけに影響している可能性もあります。
0.0022 デュシャンヌ型筋ジストロフィー
DMD、IVS26、T-G、+2
Wilton ら(1994)は、デュシャンヌ型筋ジストロフィー(DMD; 310200)の2人の兄弟から、通常よりも大きなジストロフィンmRNAをRT-PCR法で特定しました。ジストロフィン mRNA のサイズが大きくなったのは、エクソン 26/イントロン 26 接合部のスプライス部位に変異が生じ、T-to-G 置換により正常な RNA 処理が妨げられたためでした。変異の下流にある隠れたスプライス部位が処理中に活性化され、イントロン 26 の 117 塩基が組み込まれる結果となりました。この挿入により、成熟ジストロフィン mRNA にインフレームの終止コドンが導入されました。アレリック特異的検査により、Wilton ら(1994年)は、母親が変異を持っておらず、従来の検査とハプロタイプ分析によりキャリアとされた長女もDMD変異を持たないことを突き止めました。 この家族の初期のハプロタイピングは一見単純そうに見えましたが、アレリック特異的解析により性腺モザイクが明らかになったのはその後のことでした。従来の遺伝カウンセリングで疾患遺伝子座を特定するために連鎖マーカーを適用した場合、この家族では誤った結果が出たでしょう。
0.0023 デュシャンヌ型筋ジストロフィー
DMD, GLN673TER
Lenk ら (1994) は、非等量 PCR-SSCP 分析と直接シークエンス法を用いて、デュシャンヌ型筋ジストロフィー (DMD; 310200) の家族において gln673-to-ter 変異を検出しました。
Barbieri ら(1995)は独自に同様の変異を検出しました。彼らはヘテロ二重鎖分析を用いて、大きな再配列が見つからなかった40人のイタリア人DMDまたはベッカー筋ジストロフィー(BMD;300376)患者のサンプルで小規模な変異を探索しました。ヘテロ二重鎖の直接シークエンシングにより、重度のDMD表現型を持つ患者のエクソン17におけるgln673終止コドンを挿入するC-to-Tトランジションが特定されました。Muntoniら(1995年)は、この家族の一員である1人の心臓におけるジストロフィンの転写と発現に関する研究を報告しています。脳およびプルキンエ細胞アイソフォームのジストロフィンは、影響を受けた男性の筋肉で発現しているのに対し、ジストロフィンの転写および発現は心臓では認められず、唯一、遠位型Dp71ジストロフィンアイソフォームは通常心臓に存在します。43および50kDのジストロフィン関連タンパク質は、Dp71が存在するにもかかわらず、心臓では著しく減少していましたが、骨格筋では減少していませんでした。心臓におけるジストロフィンの欠如とジストロフィン結合タンパク質のダウンレギュレーションが、この家族における重度の心筋症の原因であると考えられます。Muntoniら(1995年)は、心臓におけるジストロフィン発現に対する突然変異の深刻な影響は、心臓特異的調節配列の除去による二次的なものである可能性があると推測しています。この家族は、通常は骨格筋および心筋の両方に存在する遺伝子の心臓での発現に特異的に影響する突然変異の最初の例である可能性があります。
0.0024 デュシャンヌ型筋ジストロフィー
DMD、1-BP欠失、10334CおよびIVS69、G-T、+1
PCRで増幅した産物の一本鎖構造解析を用いて、デュシャンヌ型筋ジストロフィー(DMD; 310200)またはベッカー型筋ジストロフィー(BMD; 300376)の無関係な患者20人のDMD遺伝子(エクソン60-79)の末端ドメインをスクリーニングしたTufferyらは (1995年)は、精神発達障害を伴うDMD患者2名において、2つの新規点変異を検出しました。エクソン70における1塩基対の欠失(10334delC)と、イントロン69における5’スプライス供与部位の変異(10294,+1,G-T)です。両方の突然変異はジストロフィンの早期翻訳終結を引き起こすことが予測されました。精神発達障害はDMD患者の約30%に認められます(Emery, 1993)。本研究では、Lenkら(1993)が報告した精神発達障害に関連するC末端点突然変異を持つ患者7名のうち5名に2例が追加され、認知障害に遺伝子の末端領域が関与している可能性が示唆されました。Hodgson ら (1992) は、異なる個人において、精神障害の有無にかかわらず同じ欠失が生じていること、および多くの異なる欠失が精神発達障害と関連していることを発見しました。
0025 拡張型心筋症、3B
DMD、IVS1、G-T、+1
Milasin ら(1996)は、X 連鎖性拡張型心筋症(CMD3B; 302045)の重症型家族における DMD 遺伝子のスプライス供与部位の変異について報告しています。DMD筋プロモーター、第1エクソン、およびイントロン領域の分析により、第1エクソンとイントロンの境界に1つの点突然変異が存在することが明らかになりました。この突然変異により、第1イントロンの普遍的に保存されている5′-スプライス部位のコンセンサス配列が不活性化されます。この突然変異は、GTジヌクレオチドコンセンサス配列の第1塩基のGからTへのトランスバージョンでした。この突然変異により、MseIに対する新たな制限部位が導入され、この突然変異は同一家系において疾患と共優性遺伝しました。主要なジストロフィンmRNAアイソフォーム(筋肉、脳、およびプルキンエ細胞プロモーター由来)の発現は心筋では完全に消失しましたが、脳およびプルキンエ細胞(筋肉ではない)アイソフォームは骨格筋で検出されました。抗ジストロフィン抗体を用いた免疫細胞化学的研究により、タンパク質は骨格筋では量が減少しているものの正常に分布していることが示されましたが、心筋では検出されませんでした。これらの知見から、著者は、筋ジストロフィンアイソフォームの発現が心筋機能にとって重要であることを示唆し、ジストロフィン関連拡張型心筋症における心臓の選択的障害は、心臓では代替プロモーターによるジストロフィンの代償的発現が欠如していることと関連している可能性を示唆しました。この家系の発端者は24歳の男性で、6か月前に発症した重度の心不全を訴えていました。 血清クレアチンキナーゼ値の上昇を含む骨格筋疾患の臨床的または実験室的な兆候はまったく認められませんでした。 また、この男性は数年間バスケットボールの選手として活躍していました。 さらに、この男性の弟も同じ疾患を発症していました。
0026 デュシャンヌ型筋ジストロフィー、知的発達障害、ERG b波の欠如
DMD, CYS3340TYR
デュシャンヌ型筋ジストロフィー、精神発達障害、およびERG b波の欠如(DMD; 310200)の患者において、Lenk ら(1996)は、 ジストログリカンの結合ドメインの後半部分で、システイン3340がチロシンに置換される結果となりました。 すべての筋線維で、ジストロフィンの弱い痕跡が認められました。 筋細胞膜の43-kD ベータジストログリカンの染色強度も減少していました。
0.0027 ベッカー型筋ジストロフィー
DMD、IVS2、G-T、-1
Roberts ら(1994年)は、ベッカー型筋ジストロフィー(BMD;300376)患者において、エクソン3のアクセプター部位の最初のヌクレオチドであるヌクレオチド位置301におけるG-Tトランスバージョンを報告しており、その結果、エクソン3スキップを伴う異常なスプライシングが生じます。
0.0028 中間型筋ジストロフィー
DMD、2-BP欠失、382AG
中間型筋ジストロフィー(デュシャンヌ型筋ジストロフィー(310200)とベッカー型筋ジストロフィー(300376)の中間の表現型)患者において、Roberts ら(1994)は、DMD 遺伝子のエクソン 3 の 382 番目の位置に 2 ヌクレオチド(AG)の欠失を同定し、スレオニン-58 の下流でフレームシフトを引き起こしていることを明らかにしました。
.0029 デュシャンヌ型筋ジストロフィー
DMD, GLN60TER
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Roberts ら (1994) は、DMD 遺伝子のエクソン 3 のヌクレオチド 386 における C-to-T 置換を特定し、60 位にナンセンス変異を引き起こしていることを明らかにしました。
0.0030 デュシャンヌ型筋ジストロフィー
DMD、1-BP INS、402A
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Kneppers ら (1993) は、DMD 遺伝子のエクソン 4 の 402 番目の位置に A が挿入されていることを特定しました。これにより、グルタミン酸-65 の下流でフレームシフトが起こり、タンパク質が短縮されます。
0031 デュシャンヌ型筋ジストロフィー
DMD, GLN85TER
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Roberts ら (1994) は、DMD 遺伝子のエクソン 4 のヌクレオチド 460 における C-to-T 置換を同定し、その結果、85 位に停止コドンが生じました。
0032 デュシャンヌ型筋ジストロフィー
DMD, ARG145TER
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Roberts ら(1994)はDMD遺伝子のエクソン6のヌクレオチド641におけるC-to-T置換を同定し、145番目の位置で終止コドンが生じていることを明らかにしました。
0033 ベッカー型筋ジストロフィー
DMD、ALA168ASP
ベッカー型筋ジストロフィー(BMD;300376)患者において、Roberts ら(1994)は、DMD 遺伝子のエクソン 6 のヌクレオチド 711 における C-to-A 置換を同定し、その結果、アラニン 168 がアスパラギン酸に置換しました。 .
0034 デュシャンヌ型筋ジストロフィー
DMD、1-BP欠失、724C
デュシャンヌ型筋ジストロフィー(DMD;310200)患者において、Roberts ら(1994)はエクソン6におけるヌクレオチドの欠失(724C)を同定し、ロイシン-172の下流でフレームシフトが起こり、タンパク質が短縮されることが分かりました。
.0035 ベッカー型筋ジストロフィー
DMD, TYR231ASN
ベッカー型筋ジストロフィー(BMD; 300376)患者において、Roberts ら(1994)はDMD遺伝子のエクソン8のヌクレオチド899におけるT-to-A置換を特定し、その結果、チロシン231がアスパラギンに置換しました。
0.0036 デュシャンヌ型筋ジストロフィー
DMD, GLN242TER
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Nigro ら(1992年)およびPrior ら(1994年)は、DMD遺伝子のエクソン8のヌクレオチド932におけるC-to-T置換を同定し、242番目の位置で終止コドンをコードしていることを明らかにしました。
0.0037 デュシャンヌ型筋ジストロフィー
DMD、GLU250TER
Roberts ら (1994) は、デュシャンヌ型筋ジストロフィー (DMD; 310200) の患者において、エクソン8のヌクレオチド956におけるG-to-Tトランスバージョンが報告されており、その結果、250番目の位置にストップコドンが生じ、タンパク質が短縮されています。
0.0038 デュシャンヌ型筋ジストロフィー
DMD、11-BP欠失、NT989
Roberts ら (1994) は、デュシャンヌ型筋ジストロフィー (DMD; 310200) の患者において、エクソン 8 の 989 番目の位置で 11 ヌクレオチドの欠失が起こり、スレオニン-261 の下流でフレームシフトが起こり、タンパク質が短縮されることを報告しています。 .
0039 デュシャンヌ型筋ジストロフィー
DMD, 1-BP INS, NT1554
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Kilimann ら (1992) はエクソン12の1554番目の位置にTの挿入を同定し、ロイシン-449の下流でフレームシフトが起こり、タンパク質が短縮されることが分かりました。
.0040 中間型筋ジストロフィー
DMD, GLY480TER
中間型筋ジストロフィー患者において、Prior ら(1994)はエクソン12のヌクレオチド1646におけるG-to-Tのトランスバージョンを同定し、その結果、480番目の位置にストップコドンが生じました。
.0041 デュシャンヌ型筋ジストロフィー
DMD, GLN497TER
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Lenk ら (1993) はエクソン13のヌクレオチド1697におけるC-to-T置換を同定し、497番目の位置に終止コドンをコードしています。
0.0042 ベッカー型筋ジストロフィー
BMD、IVS13、G-T、-1
ベッカー型筋ジストロフィー(BMD; 300376)患者において、萩原らは(1994年)、エクソン13の5’スプライス部位内のエクソン13の末端ヌクレオチド(1810)でG-Tトランスバージョンを同定しました。この変異によりエクソン13のスキップが生じます。予測されるポリペプチドは、バリン495から下流のアミノ酸40個が欠損した短縮ジストロフィンです。
0.0043 デュシャンヌ型筋ジストロフィー
DMD, TRP651TER
Roberts ら (1994) は、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、エクソン16のヌクレオチド2160におけるG-to-A 変異により、651番目の位置に終止コドンが生じ、タンパク質が短縮されることを報告しています。 .
0044 デュシャンヌ型筋ジストロフィー
DMD, LYS770TER
ロバーツら(1994年)は、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、エクソン19のヌクレオチド2516におけるA-to-T置換により、770番目の位置に停止コドンが生じ、タンパク質が短縮されることを報告しています。
.0045 デュシャンヌ型筋ジストロフィー
DMD, LYS773GLU
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Saad ら (1994) はエクソン 19 のヌクレオチド 2525 における A-to-G 置換を同定し、グルタミン酸をリジン-773 に変化させました。
0.0046 デュシャンヌ型筋ジストロフィー
DMD、52-bp欠失
デュシャンヌ型筋ジストロフィー(DMD;310200)患者において、松尾ら(1990年、1991年)はエクソン19の88bpのうち52bpの欠失を同定しました。エクソン19の3’末端および5’末端は存在していました。この突然変異により、エクソン20の791番目のアミノ酸残基に終止コドンが導入され、著しく短縮されたタンパク質が生成されることになりました。
0.0047 デュシャンヌ型筋ジストロフィー
DMD、1-BP INS、NT2928
Roberts ら (1994) は、デュシャンヌ型筋ジストロフィー (DMD; 310200) の患者においてエクソン21の2928番目の位置に1ヌクレオチドの挿入があり、ロイシン-907の下流でフレームシフトが起こり、タンパク質が短縮されることを報告しています。
0.0048 デュシャンヌ型筋ジストロフィー
DMD、GLN1041TER
Roberts ら (1994) は、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、エクソン23のヌクレオチド3329におけるC-to-T置換により、1041番目の位置で終止コドンが生じ、タンパク質が短縮されることを報告しています。 .
0049 デュシャンヌ型筋ジストロフィー
DMD, TRP1063TER
ロバーツら(1994年)は、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、エクソン24の3396番目のヌクレオチドでG-to-Aの変異が起こり、1063番目の位置で終止コドンが生じ、タンパク質が短縮されることを報告しています。
0.0050 デュシャンヌ型筋ジストロフィー
DMD、GLN1405TER
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Roberts ら(1994)はエクソン 30 のヌクレオチド 4421 における C-to-T 置換を同定し、1405 位に停止コドンが生じ、短縮タンパク質が生成されることを明らかにしました。 .
0051 デュシャンヌ型筋ジストロフィー
DMD, GLN1472TER
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Roberts ら (1994) はエクソン 32 のヌクレオチド 4622 における C-to-T 置換を同定し、1472 位に停止コドンが生じ、タンパク質が短縮されることを明らかにしました。
0.0052 デュシャンヌ型筋ジストロフィー
DMD, ARG1967TER
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Saad ら (1993) はエクソン 41 のヌクレオチド 6107 で C-to-T 遷移を同定し、1967 位に停止コドンをコードしていることを明らかにしました。
0.0053 デュシャンヌ型筋ジストロフィー
DMD、1-BP欠失、6408C
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Kneppers ら (1993) は1ヌクレオチドの欠失(6408delC)を同定し、スレオニン-2067の下流でフレームシフトが起こり、タンパク質が短縮されることが分かりました。.
0054 デュシャンヌ型筋ジストロフィー
DMD, ARG2098TER
ロバーツら(1994年)は、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者のDMD遺伝子のエクソン44におけるヌクレオチド6500のT-to-C置換を報告しており、これにより2098番目の位置に終止コドンが生じ、タンパク質が短縮されます。
0055 デュシャンヌ型筋ジストロフィー
DMD, GLN2125TER
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Prior ら(1993)はエクソン44のヌクレオチド6581におけるC-to-T置換を同定し、2125番目の位置に終止コドンをコードしている。
0.0056 デュシャンヌ型筋ジストロフィー
DMD、17-bp欠失、NT6982
Roberts ら(1994年)は、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者のエクソン47における6982Aから6998Cまでの17bpの欠失を特定し、グルタミン酸-2259の下流でフレームシフトが起こり、タンパク質が短縮されることを明らかにしました。
0.0057 デュシャンヌ型筋ジストロフィー
DMD、GLN2264TER
Roberts ら(1994)は、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者のエクソン 47 のヌクレオチド 6998 における C-to-T 置換を報告しており、これにより 2264 位に停止コドンが生じ、短縮タンパク質が生成されます。
0.0058 デュシャンヌ型筋ジストロフィー
DMD、1-BP INS、7188A
Roberts ら(1994年)は、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者のエクソン48の7188番目の位置に1ヌクレオチド(A)の挿入があり、グルタミン酸-2331の下流でフレームシフトが起こり、タンパク質が短縮されることを報告しています。
0.0059 デュシャンヌ型筋ジストロフィー
DMD、IVS47、G-A、+1、EX48DEL
Roberts ら (1994) は、DMD 遺伝子のエクソン 48 のドナー部位の最初のヌクレオチドであるヌクレオチド 7306 における G-to-A 遷移を報告しており、その結果、デュシャンヌ型筋ジストロフィー (DMD; 310200) の患者では異常なスプライシングが起こり、エクソン 48 がスキップされることになります。 .
0060 デュシャンヌ型筋ジストロフィー
DMD、GLU2468TER
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Winnard ら(1992)は、DMD 遺伝子の7609-10位に2ヌクレオチド置換(GGからAT)を同定し、2468位に終止コドンをコードしていることを明らかにしました。
0.0061 再分類 – 意義不明のバリアント
DMD, GLU2910VAL
このバリアントは、以前は「デュシャンヌ型筋ジストロフィー」というタイトルでしたが、Hamosh (2018) によるgnomADデータベースのレビューに基づき再分類されました。
デュシャンヌ型筋ジストロフィー(DMD;310200)患者において、Lenk ら(1994)は、DMD 遺伝子のエクソン 59 のヌクレオチド 8937 で A-to-T 置換を同定し、グルタミン酸-2910 がバリンに置換していることを明らかにしました。
Hamosh (2018) は、E2910V バリアントが gnomAD データベース(2018年4月19日)において、199,997のアレリックのうち4,287、ホモ接合型53、ヘテロ接合型1,379に存在することを明らかにしました。
0.0062 再分類 – 意義不明のバリアント
DMD, ASN2912ASP
このバリアントは、以前は「デュシャンヌ型筋ジストロフィー」というタイトルでしたが、Hamosh (2018) によるgnomADデータベースのレビューに基づき再分類されました。
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Lenk ら (1994) はエクソン 59 のヌクレオチド 8942 においてアスパラギン酸がアスパラギンに置換する A-to-G 置換を同定しました。
Hamosh (2018) は、N2912DバリアントがgnomADデータベース(2018年4月19日)において、200,085のアレリックのうち4,370、ホモ接合型59、ヘテロ接合型1,416に存在することを発見しました。
.0063 再分類 – 意義不明のバリアント
DMD, HIS2921ARG
以前は「BECKER MUSCULAR DYSTROPHY」と題されていたこのバリアントは、Hamosh (2018) によるgnomADデータベースのレビューに基づき、再分類されました。
ベッカー型筋ジストロフィー(BMD;300376)患者において、Lenk ら(1994)は、DMA 遺伝子のエクソン 59 のヌクレオチド 8970 において、ヒスチジン-2921 がアルギニンに置換する A-to-G 置換を同定しました。
Hamosh (2018) は、H292RバリアントがgnomADデータベース(2018年5月8日)において200,041のアレリックのうち5,130、ホモ接合型42、ヘミ接合型1,923に存在することを明らかにしました。
0.0064 デュシャンヌ型筋ジストロフィー
DMD, SER3066TER
ロバーツら(1994年)は、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者のDMD遺伝子のエクソン62のヌクレオチド9405におけるC-to-A置換を報告しており、これにより3066番目の位置に終止コドンが生じ、タンパク質が短縮されます。
0.0065 デュシャンヌ型筋ジストロフィー
DMD、4-BP欠失、NT9679
Roberts ら(1994)は、デュシャンヌ型筋ジストロフィー(DMD; 310200)患者のDMD遺伝子のエクソン65における9679Tから9682Tまでの4bpの欠失を報告しています。この欠失により、イソロイシン-3157の下流でフレームシフトが起こり、タンパク質が短縮されます。
0.0066 デュシャンヌ型筋ジストロフィー
DMD、IVS65、G-A、+1
Lenk ら (1993) は、デュシャンヌ型筋ジストロフィー (DMD; 310200) の患者において、エクソン 65 のドナー部位の最初のヌクレオチドであるヌクレオチド 9771 における G-to-A 変異を同定しました。その結果、異常なスプライシング、エクソン 65 のスキップ、および短縮タンパク質が生じます。
0.0067 デュシャンヌ型筋ジストロフィー
DMD, ARG3381TER
Lenk ら (1993) は、デュシャンヌ型筋ジストロフィー (DMD; 310200) の患者において、DMD 遺伝子の 10349 番目の位置で C-to-T 置換が起こり、3381 番目の位置で終止コドンがコードされていることを特定しました。
0.0068 中間型筋ジストロフィー
DMD、IVS70、G-A、+1
中間型筋ジストロフィー患者において、Lenk ら(1993)は、エクソン70のドナー部位の最初のヌクレオチドであるDMD遺伝子のヌクレオチド10431におけるG-to-Aトランジションを同定しました。その結果、リジン-3374の下流でフレームシフトが起こり、タンパク質が短縮されました。
0.0069 デュシャンヌ型筋ジストロフィー
DMD、IVS70、G-T、+5
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Lenk ら(1993)は、DMD 遺伝子のエクソン 70 のドナー部位の 5 番目のヌクレオチドであるヌクレオチド 10431 において、G-to-T 転換を同定しました。 この変異により、リジン 3374 の下流でフレームシフトが起こり、短縮型タンパク質が生じます。
.0070 ベッカー型筋ジストロフィー
DMD、ALA3421VAL
ベッカー型筋ジストロフィー(BMD; 300376)患者において、Lenk ら (1993) は、DMD 遺伝子のエクソン 71 のヌクレオチド 10470 において、アラニン 3421 がバリンに置換する C-to-T 置換を同定しました。 .
0071 ベッカー型筋ジストロフィー
DMD、1-BP欠失、10683C
ロバーツら(1994年)は、ベッカー型筋ジストロフィー(BMD;300376)患者のDMD遺伝子のエクソン74における1ヌクレオチド(10683C)の欠失を報告しており、この欠失により、アラニン-3492の下流でフレームシフトが起こり、タンパク質が短縮されることになります。
0.0072 デュシャンヌ型筋ジストロフィー
DMD、8-BP欠失、1-BP挿入、NT10692
デュシャンヌ型筋ジストロフィー(DMD; 310200)患者において、Lenk ら (1993) は DMD 遺伝子における 10692 番目の位置で 8 ヌクレオチドが G に置換していることを特定しました。その結果、ロイシン-3495 から下流へのフレームシフトが生じ、タンパク質が短縮されました。
0.0073 拡張型心筋症、3B
DMD、THR279ALA
Ortiz-Lopez ら (1997) は、X 連鎖心筋症 (CMD3B; 302045) の北米の大家族において、SSCP 分析および直接シークエンス法を用いて原因となる突然変異を特定しました。ヌクレオチド1043におけるA-to-G変異は、発症した男性および保因者のみに認められました。この変異は、ジストロフィン分子のH1領域内のアミノ酸位置279における高度に保存されたトレオニンをアラニンに変化させます。この変化は、心筋細胞膜の完全性の喪失と最終的な収縮機能の喪失をもたらすことが予測されます。
0.0074 ベッカー型筋ジストロフィー
DMD、GLU1211TER、3839G-T
志賀らは、日本人ベッカー型筋ジストロフィー患者(BMD; 300376)のDMD遺伝子第27エクソン28ヌクレオチド目におけるG-to-Tトランスバージョン(3839G-T)によるナンセンス変異、グルタミン酸1211番目の塩基がテトラロイシンに置換する変異(E1211X)を発見しました。エクソン27のスキップが部分的に生じた結果、エクソン27の両端の共通配列は変化していなかったにもかかわらず、短縮型ジストロフィンmRNAが生成されました。E1211Xがどのようにしてエクソン27のスキップを誘発するのかを解明するために、志賀らは(1997年)、HeLa細胞核抽出液を用いたキメラショウジョウバエdoublesex(dsx)遺伝子pre-mRNAを用いた試験管内スプライシングシステムにおいて、エクソン27内のプリンリッチ領域のスプライシングエンハンサー活性を調べました。3839G-Tを含む変異配列は、このキメラpre-mRNAの上流のイントロンにおける野生型プリンリッチ配列のスプライシングエンハンサー活性を消失させました。エクソン27のプリンリッチ配列を模倣した人工的なポリプリンオリゴヌクレオチドもまた、Tヌクレオチドの導入により抑制されるエンハンサー活性を示しました。さらに、挿入されたT残基によってナンセンスコドンが生じると、スプライシングエンハンサー活性はより顕著に阻害されました。これは、ナンセンス変異を有するエクソンの部分スキップが、スプライシングエンハンサー配列の破壊によるものであることを示す最初の証拠です。
0.0075 心筋症、拡張型、3B
DMD、ALU INS
X染色体遺伝性拡張型心筋症(CMD3B; 302045)の家族において、Ferliniら(1998年)は、発症した家族員が、イントロン11の5’末端から2.4kb下流のDMD遺伝子に組み込まれたAlu様移動因子を発見しました。この再配列により、イントロン11の1つの隠れたスプライシング部位が活性化され、エクソン11と12の間に挿入された、Alu様配列と隣接するイントロン11の一部を含む代替転写産物が生成されました。Alu様配列のすべてのフレームに多数の終止コドンが存在するため、代替転写産物の翻訳は途中で途切れます。心筋では変異型 mRNA のみが検出されましたが、骨格筋では正常型 mRNA と共存していました。Ferlini ら (1998) は、この Alu 様配列は、転移性因子として知られる因子と繰り返し、クラスター化し、トランスポジションを起こす可能性のある、新規の反復配列である可能性を示唆しています。
0.0076 デュシャンヌ型筋ジストロフィー
DMD, ARG3190TER
デュシャンヌ型筋ジストロフィー(DMD;310200)の9.5歳男性で、知能テストが実施できず、読解能力もなかった患者について、Moizard et al.(2000)はDp71転写物の早期翻訳終結を特定しました。エクソン66における9776C-Tの変異により、arg3190が停止置換されました。
0.0077 ベッカー型筋ジストロフィー
DMD, ARG1314TER
Ginjaar ら (2000) は、3人の男性に異なる表現型が現れたX連鎖筋ジストロフィーの家族を研究しました。その3人とは、心筋症を伴う重症のベッカー型筋ジストロフィー患者(BMD; 300376)、軽症のBMD患者、および血清クレアチンキナーゼ値の上昇が認められる一見健康な男性です。軽症患者の筋生検標本では、4つの抗体(NCL-DYS1)のうち1つがジストロフィンの欠如を示しました。タンパク質欠失試験では、この患者の筋組織とリンパ球増多の両方で、欠失ジストロフィンが検出されました。ゲノム配列解析では、DMD遺伝子のエクソン29に4148C-T変異が認められ、アルギニン1314がセリン1314に変異していました。より大きなペプチドのmRNA断片の配列解析では、エクソン29のスキップが示され、オープンリーディングフレームが復元されました。したがって、抗体NCL-DYS1のエピトープはエクソン29に位置します。3人の男性の臨床症状が健康な状態から重度の症状まで様々であることは、エクソン29のスキップのレベルに関連しているようです。この発見は、DMDにおけるエクソン・スキップを誘導し、より軽度の疾患を生み出すことを目的とした遺伝子治療戦略の将来性を強調するものです。
0.0078 デュシャンヌ型筋ジストロフィー
DMD、1-BP欠失、377A
デュシャンヌ型筋ジストロフィー(DMD;310200)の男性患者において、van Essen ら(2003年)はDMD遺伝子における1塩基対欠失(377delA)を特定しました。この欠失によりフレームシフトが起こり、45塩基対下流のコドン141で終止コドンが生じました。母親は変異に対してモザイク状態であり、もう一人の息子には「リスクハプロタイプ」が受け継がれましたが、その息子は健康でした。
0.0079 デュシャンヌ型筋ジストロフィー
DMD、16-bp欠失
重度のデュシャンヌ型筋ジストロフィー(DMD;310200)患者において、Todorova ら(2003)は、DMD 遺伝子のエクソン 44 に 16-bp の欠失を同定しました。この欠失は、フレームシフトと早期終止翻訳につながりました。この患者は10歳で車椅子生活を余儀なくされました。14歳で心筋障害が検出され、18歳で心筋症により死亡しました。 .
0080 ベッカー型筋ジストロフィー
DMD, IVS62, A-G, -285
ベッカー型筋ジストロフィー(BMD)の無関係な患者2人(300376)において、Tuffery-Giraud ら(2003)はDMD遺伝子に2つの深層イントロン変異を同定し、成熟転写物における偽エクソンの異常な挿入を引き起こしていることを明らかにしました。この2つの変異は、筋肉から単離した転写産物に対してRT-PCR法を用いて特定されました。エクソン62とエクソン63の間の58塩基対の挿入により生じた最初の異常な転写産物は、精神発達障害を伴うBMD患者で特定されました。この転写産物の起源は、高品質のドナースプライシング部位の発生につながったイントロン62の変異(IVS62-285A-G)でした。2つ目の変異であるIVS25+2036A-G(300377.0081)は、イントロン25に存在し、クレアチンキナーゼ値が高い潜在性BMD患者で確認されました。この変異は、既存のアクセプター・スプライス部位の強度を強化し、結果として95bpのイントロン性偽エクソンの活性化につながりました。変性高速液体クロマトグラフィー(DHPLC)を使用して、患者の母親が体細胞モザイクであることが判明しました。新たに確認されたエクソン挿入により早期終止コドンが生じましたが、両患者ではある程度の正常なスプライシングが行われていました。
.0081 ベッカー型筋ジストロフィー
DMD, IVS25, A-G, +2036
300377.0080およびTuffery-Giraudら(2003年)を参照のこと。
.0082 デュシャンヌ型筋ジストロフィー
DMD, ARG2905TER
デュシャンヌ型筋ジストロフィー(DMD)患者141人のうち6人(310200)で、DMD遺伝子における大きな欠失が陰性であることが以前に判明していたが、Buzin et al.(2005年)は DMD遺伝子のエクソン59のCpGジヌクレオチドにおける8713C-Tトランジションを特定し、その結果、アルギニン2905がテトラロイシン(R2905X)に置換しました。ハプロタイプ分析により、6つの変異はすべて独立して発生したことが示されました。 .
0083 ベッカー型筋ジストロフィー、非定型
DMD, IVS2, T-A, +5591
無症候性ジストロフィノパチー(300376参照)の12歳の男児において、Yagiら(2003)は、リンパ球増多および筋mRNAの両方において、DMD遺伝子のエクソン2とエクソン3の間に132bpの挿入があることを観察しました。挿入部位の隣接領域の配列を決定したところ、IVS2に+5591T-Aのトランスバージョンが発見され、ジストロフィンmRNAに組み込まれる新規エクソン構造を生み出すと予測されるスプライシングアクセプター部位のAGジヌクレオチドのコンセンサス配列が形成されました。44コドン配列は、おそらくジストロフィンの局在を妨害すると思われるアクチン結合部位の間に挿入されています。この患者の筋生検の免疫組織化学染色により、N末端ドメインは染色されず、ロッドおよびC末端ドメインは不連続で斑状に染色されていることが明らかになりました。Yagi ら(2003年)は、エクソン構造の余分なエクソンにつながる一塩基の変化によるスプライスアクセプター部位の生成は、ヒト疾患における新規の分子メカニズムであると述べています。
0.0084 デュシャンヌ型筋ジストロフィー
DMD、TYR1995TER
重度のデュシャンヌ型筋ジストロフィー(DMD; 310200)の日本人男児において、Tran ら(2007)は、DMD 遺伝子のエクソン 42 における 5985T-G 転移を同定しました。これにより、tyr1995 が ter(Y1995X)に置換され、機能しないタンパク質が生じます。患者の骨格筋のRNA分析では、短縮型転写物の独占的発現が認められましたが、患者の白血球のRNA分析では、5985T-Gの変異が新たなスプライスアクセプター部位も作り出し、その結果、異常なスプライシングが起こり、部分的に機能するジストロフィンタンパク質が2種類生成されることが分かりました。この発見は、組織特異的なスプライシング制御を示唆しており、これはシスエレメントよりもトランスエレメントによるものである可能性が高いと考えられます。Tran ら(2007年)は、このような症例におけるDMDスプライシングの調節が、潜在的な治療標的となりうると示唆しています。 .
0085 ベッカー型筋ジストロフィー、非定型
DMD、EX45-47DEL
Kerst ら(2000年)は、MYF6遺伝子における387G-Tトランスバージョンによるヘテロ接合不全が、ala112-to-ser変異(159991.0001)を引き起こし、軽度の中心核ミオパチー(160150)の原因となることを発見しました。発端者の父親にも同じ変異があり、DMD遺伝子のエクソン45から47の欠失も認められたため、ベッカー型筋ジストロフィー(BMD; 300376)の軽症型から重症型への転換が起こりました。父親は21歳で車椅子生活を余儀なくされました。Kerst ら (2000) は、DMD遺伝子におけるエクソン45から47の欠失は、軽度から中程度のBMDの経過と関連していることが知られていると指摘しています。 .
0086 ベッカー型筋ジストロフィー
DMD、TRP3TER
ユタ州在住の58歳男性のベッカー型筋ジストロフィー(BMD; 300376)の軽症例において、Flanigan ら(2003)はDMD遺伝子のエクソン1に9G-Aトランジションを同定し、その結果、trp3-to-ter(W3X)置換が生じました。この患者は20歳で筋力低下を発症しましたが、58歳になっても歩行能力を失うことはありませんでした。Gurvich ら(2009年)は、Flanigan ら(2003年)が報告した患者の追跡調査を行い、その患者は62歳で歩行能力を失いました。この患者の兄弟も同じ変異を持っていましたが、62歳になっても階段の昇り降りに若干の困難を感じる程度でした。Gurvich ら(2009年)は、最初の20年間で血清クレアチンキナーゼの増加や時に労作性筋痛が現れる軽度の骨密度減少症の患者5人家族に、W3X変異を特定しました。SNP解析により、ユタ州の住民集団に創始者効果があることが示され、この対立遺伝子の維持は、非常に軽度の表現型による可能性が高いことが示唆されました。
Gurvich ら(2009年)は免疫蛍光法を用いて、W3X 変異を持つ患者の筋線維ではジストロフィンタンパク質が発現していることを発見しました。ただし、そのレベルは対照群と比較して低下しており、W3X 変異タンパク質はエクソン 1 および N 末端配列を欠いていました。DMDの筋肉アイソフォームのエクソン1から9までを網羅するレポーター構築物を用いた研究により、DMD遺伝子のエクソン6に2つの翻訳開始部位が存在することが確認され、その結果、野生型の72kDタンパク質ではなく、60kDのタンパク質が合成されることが分かりました。この発見は、この結果生じた短縮タンパク質が、DMD遺伝子のエクソン1における欠失変異によって生じると予想される重度の表現型を大幅に改善するのに十分な残存活性を保持していることを示唆しています。







