承認済シンボル:DLD
遺伝子名:dihydrolipoamide dehydrogenase
参照:
HGNC: 2898
AllianceGenome : HGNC : 2898
NCBI:1738
Ensembl :ENSG00000091140
UCSC : DLD (ENST00000205402.10) from GENCODE V46
遺伝子OMIM番号238331
●遺伝子のlocus type :タンパク質をコードする
●遺伝子のグループ:Pyruvate dehydrogenase complex
Flavoproteins
Oxoglutarate dehydrogenase complex
●遺伝子座: 2898
●ゲノム座標:7:107,891,107-107,921,198
遺伝子の別名
diaphorase
dihydrolipoamide dehydrogenase (E3 component of pyruvate dehydrogenase complex, 2-oxo-glutarate complex, branched chain keto acid dehydrogenase complex)
dihydrolipoyl dehydrogenase
DLDH
DLDH_HUMAN
E3 component of pyruvate dehydrogenase
GCSL
glycine cleavage system L protein
LAD
lipoamide dehydrogenase
lipoamide reductase
lipoamide reductase (NADH)
lipoyl dehydrogenase
PHE3
pyruvate dehydrogenase component E3
遺伝子の概要
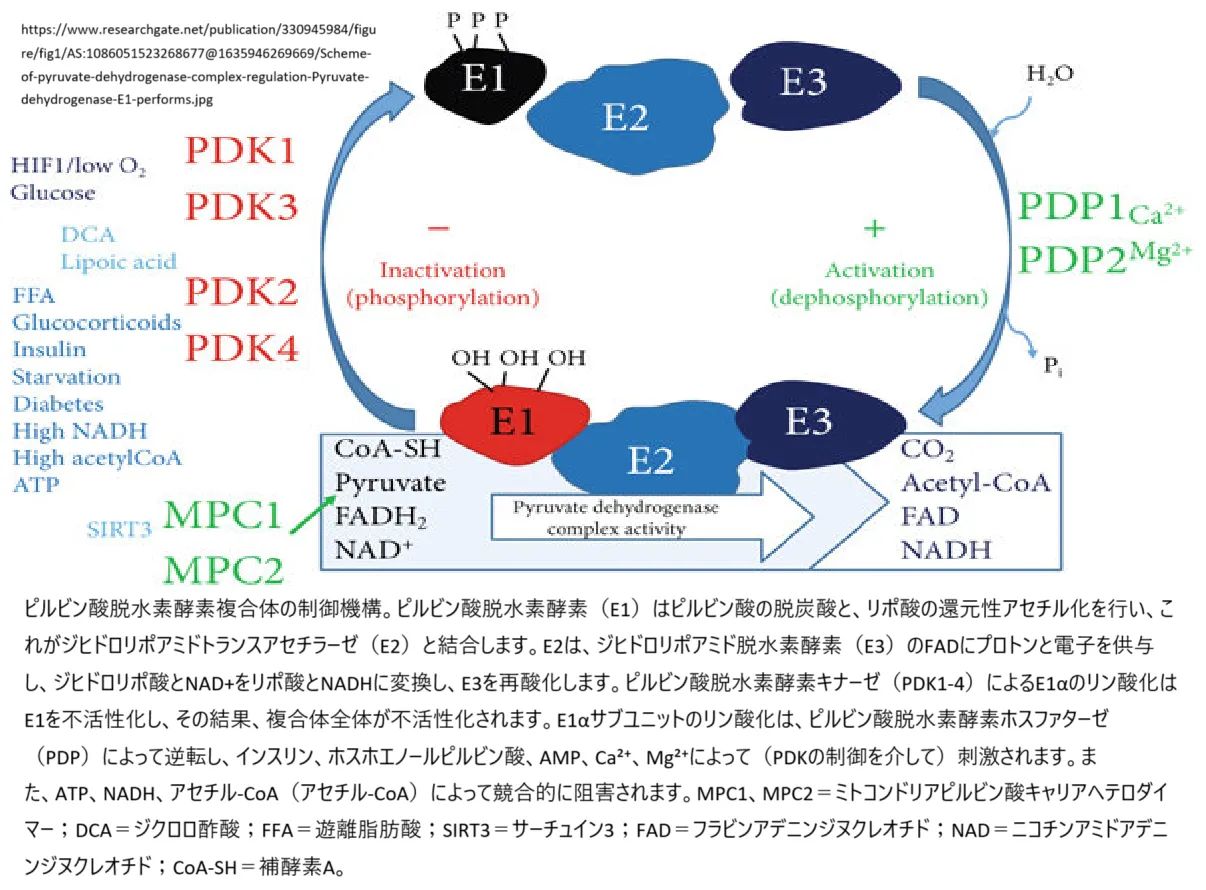
1. ピルビン酸脱水素酵素複合体(PDC)
– 炭水化物が代謝される際、ピルビン酸をアセチルCoAに変換するために働く複合体です。これにより、エネルギー生成の主要経路であるクエン酸回路が進行します。
2. α-ケトグルタル酸脱水素酵素複合体(KGDC)
– クエン酸回路の一部で、α-ケトグルタル酸をスクシニルCoAに変換し、エネルギー生成に寄与します。
3. 分岐鎖α-ケト酸脱水素酵素複合体(BCKDC)
– 分枝鎖アミノ酸(ロイシン、イソロイシン、バリン)の分解に関与し、これらのアミノ酸をエネルギー源に変換します。
酵素の構造と機能
– DLD酵素は、ホモダイマー(二量体)として機能し、E2成分(DBTタンパク質)に共有結合しているリポ酸補酵素を酸化的に再生し、NAD+をNADHに変換する触媒作用を持っています。これにより、エネルギー代謝に必要なNADHが生成されます。
その他の機能
– DLD酵素は、ミトコンドリアグリシン開裂系(GCS)の一部でもあり、この系では「Lタンパク質」とも呼ばれます。GCSは、グリシンというアミノ酸の代謝に重要です。
– さらに、ダイアフォラーゼおよびセリンプロテアーゼとしての「隠れた活性」も持っており、これらの機能により、さまざまな代謝過程にも関与していることが示唆されています。
まとめ
DLD遺伝子は、エネルギー代謝において重要な役割を果たす多機能な酵素を作り出す指示を提供します。PDC、KGDC、BCKDCの3つの主要な酵素複合体に共通のE3成分として働き、NADH生成を促進します。また、ミトコンドリア内でグリシンの分解にも関わり、他の潜在的な活性も持つ、非常に重要な酵素です。
遺伝子の発現とクローニング
さらに、Ponsら(1988年)は、ヒト肝臓のcDNAライブラリーから、ジヒドロリポアミド脱水素酵素の全コード領域を含むcDNAクローンを単離しました。この配列に基づき、509個のアミノ酸から成る前駆体タンパク質がコードされていることが確認されました。成熟型のE3酵素はホモダイマー(二量体)であり、各サブユニットの分子量は約50kDaです。
また、ブロットハイブリダイゼーション分析を用いて、ヒトの組織や線維芽細胞において2.2kbおよび2.4kbの2種類のmRNA転写物が検出されました。これにより、ジヒドロリポアミド脱水素酵素が複数の転写物を持ち、広範な組織で発現していることが確認されています。
遺伝子の構造
この発見は、ジヒドロリポアミドデヒドロゲナーゼ(E3)酵素の生成に関与する遺伝子構造を理解するための重要なステップです。エクソンは、RNAスプライシングの過程でmRNAに組み込まれ、最終的なタンパク質のアミノ酸配列に寄与するため、これらのエクソンの構成は酵素の正しい機能に直接影響します。
マッピング
さらに、Schererら(1991年)は、より詳細な解析を行い、E3遺伝子の位置を7q31-q32という特定の領域に修正しました。この領域は、ヒト第7染色体の長腕(q腕)に位置しており、エネルギー代謝に関わる重要な遺伝子が集まる領域の一部です。
また、Johnsonら(1997年)は、種間戻し交配分析(異なる種間での交配を使って遺伝子の位置を特定する手法)を用いて、マウスのDld遺伝子を12番染色体の近位領域にマッピングしました。この領域は、ヒトの7q31-q32に相当する領域であり、マウスとヒトの間でこの遺伝子が進化的に保存されていることを示しています。
遺伝子の機能
● DLDの役割と新たな発見
1. 鉄代謝と活性酸素制御
Petratら(2003年)は、DLDがNAD(P)Hを使って鉄錯体を1電子還元することで、細胞内の不安定鉄プールに影響を与えることを発見しました。このプロセスは、鉄依存性の活性酸素種(ROS)生成による細胞毒性に影響を与える可能性があります。これは、細胞の鉄の運搬や代謝に重要な役割を果たすと考えられます。
2. DLDのプロテアーゼ活性
Babadyら(2007年)は、DLDがホモダイマーとして安定しているときには通常の酵素機能を果たしますが、単量体(モノマー)になるとセリンプロテアーゼとして機能することを発見しました。特定の残基(S456とE431)がプロテアーゼ活性の中心であり、これらが変異すると、その活性が失われます。DLDは、ミトコンドリアタンパク質であるフラタキシン(FXN)のN末端から重要なドメインを除去し、これが鉄代謝と抗酸化保護に関連する役割を持ちます。フラタキシンに異常があると、神経変性疾患であるフリードライヒ失調症(FRDA)を引き起こします。
3. 酵素活性と酸化還元反応
DLDは、通常の順反応ではジヒドロリポ酸を酸化し、NAD+からNADHを生成します。一方、逆反応ではリポ酸などの基質を還元し、NADHをNAD+に酸化します。もしNAD+やリポ酸が不足すると、酸素がスーパーオキシドアニオンに還元され、活性酸素種(ROS)が生成されます。これにより、DLDはダイアフォラーゼとして機能し、1電子を有機分子に伝達することで還元反応を行います。また、DLDがモノマー化すると、このダイアフォラーゼ活性が増加することが考えられています。
● まとめ
– Lタンパク質(DLD)は、グリシン開裂系や他の重要な酵素複合体の一部であり、鉄代謝や活性酸素制御に関わる。
– DLDのモノマー化は、セリンプロテアーゼやダイアフォラーゼとしての新たな機能を発揮し、特定のミトコンドリアタンパク質の分解や活性酸素の生成に影響を与える。
– フラタキシンとの相互作用が、フリードライヒ失調症のような疾患に関連している。
分子遺伝学
1. ジヒドロリポアミド脱水素酵素欠損症とDLD遺伝子変異
– 坂口ら(1986年)によるDLDD患者の報告後、劉ら(1993年)は、DLD遺伝子のミスセンス変異(238331.0001および238331.0002)による複合ヘテロ接合性を確認しました。
– Shaagら(1999年)は、アシュケナージ系ユダヤ人におけるLAD欠損症患者の多くに、DLD遺伝子のG229C変異(238331.0006)が存在することを発見しました。ホモ接合性のG229C変異は、比較的軽度の症状と関連していました。
– Grafakouら(2003年)は、Leigh症候群の患者において、DLD遺伝子に新規変異(238331.0007および238331.0008)があることを確認しました。
2. DLD変異と酵素活性の影響
– Babadyら(2007年)は、DLDホモダイマー界面にあるいくつかの変異が、セリンプロテアーゼ活性の増強とともに、DLD活性の部分的または完全な喪失を引き起こすことを発見しました。これにより、代謝活性が失われる一方でプロテアーゼ活性が増加するというメカニズムが示唆されています。
3. DLD変異と活性酸素種(ROS)の生成
– Ambrus氏ら(2011年)は、大腸菌で精製したDLD変異体を用いたin vitro(試験管内)解析で、D479V、E375K、P488L、G194Cなどの変異が活性酸素種(ROS)の生成速度を大幅に増加させることを発見しました。これらの変異により、酸化ストレスに対する感受性も増加しました。
4. DLD変異による疾患表現型の多様性
– Vaubel氏ら(2011年)の研究では、DLDの二量体界面に変異があると、酵素のジアホラーゼ活性やタンパク分解活性が強化され、多臓器障害や呼吸機能の低下が進行することが示されました。特に、E375KやD479Vなどの変異は重症の表現型と関連していました。
– 酵母細胞での解析でも、DLDの残存活性がわずかでも、酸化ストレスが高まることで、リポ酸補酵素やミトコンドリアタンパク質に対する損傷が加速されました。
5. 臨床的重症度に対するDLD変異の影響
– Vaubelら(2011年)は、DLDの隠れた活性が酸化損傷を促進し、DLD変異の影響を受けた患者の臨床的重症度のばらつきに寄与している可能性を示唆しました。これにより、DLD欠損に関連する疾患の治療において、抗酸化物質の使用が有益である可能性も示されています。
まとめ
DLD遺伝子の変異は、酵素活性の低下や活性酸素種の増加、さらには代謝プロセスの異常を引き起こし、複数の疾患や表現型に関連しています。特に、二量体界面や酸化ストレスとの関わりが、臨床的な重症度に大きく影響することがわかっています。
アレリックバリアント
DLD、LYS72GLU
坂口ら(1986年)により報告されたジヒドロリポアミドデヒドロゲナーゼ欠損症(DLDD; 246900)患者において、劉ら(1993年)はDLD遺伝子におけるミスセンス変異の複合ヘテロ接合性を示しました。A-Gの変化により、リジン72がグルタミン酸(K72E (K37E、Odievre et al., 2005)、およびC-T変化によるpro488-to-leu(P488L; 238331.0002)置換(P453L、Odievre et al., 2005)が起こります。これらの変異は活性部位を変化させ、おそらくはFADの結合にも影響を与えています。
Ambrusら(2011年)は、in vitroでの機能発現研究により、P453L変異はLADH活性の著しい低下と活性酸素種の生成の著しい増加をもたらすことを明らかにしました。.
0002 ジヒドロリポアミドデヒドロゲナーゼ欠損症
DLD、PRO488LEU
ジヒドロリポアミド脱水素酵素欠損症(DLDD; 246900)患者で発見されたDLD遺伝子のpro488-to-leu(P488L)変異に関する議論については、Liuら(1993年)の238331.0001を参照してください。
.0003 ジヒドロリポアミド脱水素酵素欠損症
DLD、1-BP INS、105A
Craigen(1996)により最初に報告されたジヒドロリポアミドデヒドロゲナーゼ欠損症(DLDD;246900)の患者について、Hong ら(1996)は、DLD 遺伝子における2つの変異による複合ヘテロ接合性を特定しました。リーダー配列の最後のコドンにおける1bpの挿入(105insA)は、フレームシフトと早期終結(Y35X)を引き起こすことが予測され、アルギニン495がグリシンに置換する(R495G;238331.0004)変異(処理されたタンパク質ではR460G、Odievre et al.、2005年)が認められました。この患者は発育遅延、低緊張、代謝性アシドーシス(血清乳酸およびピルビン酸の上昇)、一過性新生児低血糖症の既往歴、およびリー症候群の特徴を示し、28ヵ月で死亡しました。患者の血漿アミノ酸分析では当初、ロイシン、イソロイシン、バリンの増加が認められました。尿有機酸分析では、乳酸、2-ヒドロキシ酪酸、3-ヒドロキシ酪酸、α-ケトグルタル酸、および3-ヒドロキシイソ吉草酸が軽度から中等度に増加していました。 28ヶ月で死亡しました。 患者リンパ球増多におけるPDCおよびE3の活性は、それぞれ対照値の26%および2%であり、患者線維芽細胞ではそれぞれ11%および14%でした。線維芽細胞における KGDC 活性は 20% でした。臨床的に異常のない両親における対応する値は、KGDC を除いて正常値の約 50% でした。これらの知見は、E3 の部分的な減少は線維芽細胞における KGDC 活性の律速段階ではないことを示唆しています。また、患者ではグリシンも増加していませんでした。
DLDDを有するアシュケナージ系ユダヤ人の血統を持つ2人の患者において、Elpelegら(1997年)はDLD遺伝子における105insA突然変異を特定しました。2人の患者は、この突然変異のヘテロ接合型であり、コード領域では他の突然変異は特定されませんでした。105insA変異のヘテロ接合性は、患者1の父親と兄弟1人のcDNA、および患者2の母親と姉妹1人のcDNAでも確認されました。両患者の筋肉組織におけるリポアミド脱水素酵素の酵素活性は、対照群の平均値の8~20%にまで低下していたため、Elpelegら(1997年)は、両患者は、この変異と別の未同定の変異による複合ヘテロ接合型であると推定しました。.
0004 ジヒドロリポアミド脱水素酵素欠損症
DLD、ARG495GLY
Hongら(1996年)により複合ヘテロ接合状態で発見されたジヒドロリポアミドデヒドロゲナーゼ欠損症(DLDD; 246900)患者のDLD遺伝子におけるarg495-to-gly(R495G)突然変異に関する考察については、238331.0003を参照してください。
.0005 データベースから削除.
0006 ジヒドロリポアミド脱水素酵素欠損症
DLD、GLY229CYS G229C変異は、14のアレリック変異のうち12を占めていました。G229Cは成熟タンパク質ではG194Cに相当します(Odievre et al., 2005)。アシュケナージ系ユダヤ人の匿名の845人のサンプルでは、G194C変異のヘテロ接合体が9例確認され、キャリア率は1:94でした。この系列の他の2つのアレリックには、以前に同定された挿入変異(238331.0003)がありました。 疾患の経過および発症年齢は、非常に多様でした。 患者の中には神経学的後遺症がほとんどなく、長生きする人もいました。 2人の患者は出生直後に発症し、9人は2歳頃、2人は成人してから発症しました。 すべての患者に嘔吐、腹痛、肝腫大の再発性エピソードが見られ、通常、エピソード中は神経学的徴候も伴いました。発作は乳酸アシドーシス、異常な肝酵素、およびプロトロンビン時間の延長を伴っていました。分枝鎖アミノ酸やα-ケト酸の増加などの生化学的異常はあまり見られませんでした。新生児期に発症した2人の患者は、注意欠陥・多動性障害、軽度の運動失調、運動協調障害、筋緊張低下、筋力低下などの神経学的障害が残りました。幼児期または成人期に発症した9人の患者は、代償不全のエピソードの間に労作性疲労に苦しみましたが、それ以外では無症状で、精神運動発達は正常でした。2人の患者は難治性の代謝性アシドーシスと多臓器不全により死亡しました。すべての患者において、E3活性は筋肉またはリンパ球増多において、コントロール値の8~21%に減少していました。検査した4人の患者では、筋肉内のE3タンパク質が対照の20~60%に減少していました。
Hongら(2003年)は、近親婚のパレスチナ系アラブ人イスラム教徒の両親から生まれた2人の同胞について報告しています。この2人は、G194C変異のホモ接合性によるE3欠損症でした。女児は、嘔吐を繰り返す脳症のエピソード中に乳児期に死亡しました。以前にも2人の兄弟が同様の状況で死亡しています。弟は生後8ヶ月から、脳症に伴う嘔吐を繰り返していました。10歳時の検査では、全身の筋力低下と消耗、運動失調性歩行、肝腫大、乳酸アシドーシスが認められました。リボフラビン、コエンザイムQ、ビオチン、カルニチンによる治療が行われました。6年後、彼は通常の学校でうまく機能していましたが、軽度の運動失調症と意思痙攣がありました。 2人の患者は、アシュケナージ系ユダヤ人の家系で、E3欠損症の重症型とG194C変異がありました。 1人は低血糖症の反復発作があり、4歳で持続性植物状態となり、まもなく死亡しました。幼児期から嘔吐とアシドーシスを繰り返していた女児は、5歳で肝不全により死亡しました。すべての患者でE3タンパク質のレベルが低下しており(対照群の35~68%)、E3活性も低下していました(対照群の8~33%)。Hongら(2003年)は、リボフラビンと追加のサプリメントで治療した1人の子供に良好な結果が得られたことを強調しています。
Ambrus ら(2011年)は、in vitro 機能発現研究により、G194C 変異タンパク質は LADH 活性に有意な変化をもたらさないが、活性酸素種の生成を有意に増加させることを明らかにしました。
0.0007 ジヒドロリポアミド脱水素酵素欠損症
DLD、ILE393THR
ジヒドロリポアミド脱水素酵素欠損症(DLDD;246900)の患者において、Grafakou ら(2003年)は、DLD 遺伝子における2つの変異のヘテロ接合性を特定しました。 エクソン11におけるI393T置換(タンパク質の二量体形成を妨げると推定)と、共通スプライス部位(238331.0008)におけるIVS9+1G-A変化です。I393Tは成熟タンパク質におけるI358Tに対応します(Odievre et al., 2005)。I358T 変異は、エクソン13の多型である1422A-Cと共優性であり、cDNA研究では両者ともホモ接合型であるように見え、スプライス部位の変異によるmRNA産物が不安定であることを示唆しています。 発症から1年後、患者は脳卒中のような発作を起こし、脳MRIでは、リー症候群に一致する対称性の高信号が認められました(256000)。
0008 ジヒドロリポアミド脱水素酵素欠損症
DLD、IVS9DS、G-A、+1
Grafakou ら(2003年)によりジヒドロリポアミド脱水素酵素欠損症(DLDD; 246900)患者において複合ヘテロ接合状態で発見された DLD 遺伝子(IVS9+1G-A)のスプライス部位変異に関する考察については、238331.0007を参照してください。
.0009 ジヒドロリポアミド脱水素酵素欠損症
ジヒドロリポアミドデヒドロゲナーゼ欠損症(DLDD; 246900)の10週齢の男児において、Cerna ら (2001) は、DLD 遺伝子における2つの変異による複合ヘテロ接合性を特定しました。 グルタミン酸375がリジンに置換する1123G-Aの変異と、メチオニン361がバリンに置換する1081A-Gの変異(M361V; 238331.0010)が確認されました。E375KとM361Vは、それぞれ成熟タンパク質のE340KとM326Vに対応します(Odievre et al., 2005)。患者のリンパ球増多、筋ミトコンドリア、および線維芽細胞におけるDLD活性は、コントロール値の5%未満であり、ウェスタンブロット分析では、DLDタンパク質レベルがコントロールの40%に減少していることが示されました。
Cameron ら(2006年)は、E375K 変異が DLD タンパク質の中央ドメインの保存されたアミノ酸残基で起こることを指摘しています。.
0010 ジヒドロリポアミドデヒドロゲナーゼ欠損症
DLD, MET361VAL
Cerna ら (2001) によりジヒドロリポアミドデヒドロゲナーゼ欠損症 (DLDD; 246900) の患者において複合ヘテロ接合型で発見された DLD 遺伝子の変異である、met361-to-val (M361V) 変異については、238331.0009 を参照してください。
.0011 ジヒドロリポアミド脱水素酵素欠損症
DLD、ASP479VAL
Shany ら (1999) は、重度の神経変性型ジヒドロリポアミド脱水素酵素欠損症 (DLDD; 246900) のイスラム系9ヶ月女児において、DLD遺伝子におけるホモ接合型14 DLD遺伝子における1436A-Tのホモ接合型トランスバージョンが確認され、asp479-to-val(D479V)置換が起こっていることが判明しました。これは、処理されたタンパク質ではD444Vの変化です(Odievre et al., 2005)。この置換は、DLD二量体の界面ドメインで起こっており、ホモ二量体の安定性を乱すことが推測されています。Shany ら (1999) が報告した患者は、生後3日目に無関心、摂食不良、および嗜眠状態を示しました。検査室での研究では低血糖症と重度の乳酸アシドーシスが認められましたが、分枝鎖ケトン酸とαケトグルタル酸は正常値を示しました。筋生検ではピルビン酸脱水素酵素複合体の活性は認められず、αケトグルタル酸脱水素酵素複合体の活性は重度に低下(2%)し、DLD活性は対照群の15%に減少していました。両親はそれぞれDLDタンパク質の活性が約50%低下していました。彼女は代謝性アシドーシスの再発を繰り返し、その多くは感染症が誘因となっていました。臨床症状には、小頭症、精神運動発達の遅れ、失明、難聴、低緊張、鋭敏な反射、軽度の肥大型心筋症が含まれます。Shany ら (1999) は、G229C 変異 (238331.0006) はさらに低い活性 (7%) と関連しているものの、より軽度の表現型であるため、この患者の表現型の重症度は残存する DLD タンパク質の活性とは相関しないと指摘しています。
Babady ら(2007年)は、D444V 変異タンパク質は野生型と比較してタンパク質分解活性が増加しており、このタンパク質分解活性は変異型 DLD タンパク質のモノマー分画と相関していることを発見しました。このタンパク質分解活性の出現が、表現型の重症化に寄与している可能性があります。
0.0012 ジヒドロリポアミド脱水素酵素欠損症
DLD, ARG482GLY
近親婚の親から生まれたアルジェリア人の3人兄弟姉妹で、ジヒドロリポアミド脱水素酵素欠損症(DLDD; 246900)を発症した症例において、Odievre ら(2005年)は、 DLD遺伝子のエクソン13における1444A-Gのホモ接合型変異が同定され、その結果、ジマー界面ドメインにおいてアルギニン482がグリシンに置換(R482G)(処理されたタンパク質ではR447G)されました。患者は、Bonnefont ら(1992年)により当初、α-ケトグルタル酸脱水素酵素欠損症と報告されていました。 患者は重度の症状を示し、全員が30ヶ月齢までに死亡しました。
0.0013 ジヒドロリポアミド脱水素酵素欠損症
DLD、ILE47THR
ジヒドロリポアミド脱水素酵素欠損症(DLDD; 246900)の2人の2等親アシュケナージ系ユダヤ人の患者において、Cameron ら(2006年)はDLD遺伝子における2つの変異の複合ヘテロ接合性を特定しました。両患者ともエクソン3にヘテロ接合性のT-to-C転移があり、FAD機能ドメイン内の高度に保存された領域で、イレ47がスレ(I47T)に置換していました。一方の患者はもう一方のアレリックに G229C (238331.0006) を有し、もう一方の患者はもう一方のアレリックに E375K (238331.0009) を有していました。 発症していない両親は、いずれもこれらの突然変異のヘテロ接合型でした。両患者とも、KGDH(それぞれ25%および44%)およびBCKDH(それぞれ58%および62%)複合体の活性が低下していましたが、PDH複合体の活性は正常範囲の下限でした(それぞれ69%および59%)。DLD活性は両患者で低下していました。G229C変異を有する患者は、その変異を有さない患者と比較して、より軽度の表現型を示しました。
| 遺伝子 | DLD |
| 疾患名 | メープルシロップ尿症3型 |
| スーパーNIPTジーンプラスで検査対象のバリアント | c.199-1G>A c.214A>G c.685G>T c.1123G>A c.1178T>C c.1236+1G>T c.1436A>T c.1444A>G c.1463C>T c.1483A>G |
| 検出率 | >85% |
| 分布 | アシュケナージ系ユダヤ |
| 引用 | Scott, S. A. et al. (2010); Quinonez, S. C. & Thoene, J. G. (2014); Shaag, A. et al. (1999) |
| 程度 | 重度 |
| 遺伝形式 | 常染色体劣性 |
| 症状:引用元 | www.nanbyou.or.jp/entry/4814 |
| 症状 | 新生児期発症の急性期では元気がない、哺乳力低下、不機嫌、嘔吐などで発症する。進行すると意識障害、けいれん、呼吸困難、筋緊張低下、後弓反張などが出現し、治療が遅れると死亡するか重篤な神経後遺症をのこす。慢性症状としては発達障害、精神運動発達遅滞、失調症、けいれんなどがみられる。 |
| 頻度 | |
| 保因者頻度 | |
| 新生児マススクリーニング | 対象 |
↑↑↑↑↑↑ テーブルこの上にペースト ↑↑↑↑↑↑







