承認済シンボル:CTNS
遺伝子名:cystinosin, lysosomal cystine transporter
参照:
HGNC: 2518
AllianceGenome : HGNC : 2518
NCBI:1497
Ensembl :ENSG00000040531
UCSC : uc002fwb.4
遺伝子OMIM番号606272
●遺伝子のlocus type :タンパク質をコードする
●遺伝子のグループ:Solute carrier family 66
●遺伝子座: 17p13.2
●ゲノム座標:(GRCh38): 17:3,636,459-3,663,103
遺伝子の別名
CTNS_HUMAN
Cystinosis
PQLC4
遺伝子の概要
CTNS遺伝子は、7回膜貫通ドメインを持つタンパク質をコードしており、その機能はリソソームからのシスチン輸送に特化しています。このタンパク質の活性は、リソソーム膜のH+(プロトン)電気化学的勾配によって駆動されます。CTNS遺伝子の変異はシスチン症の発症につながり、選択的スプライシングによって複数の転写産物が生じます。シスチン症は、リソソーム内にシスチンが蓄積することで特徴づけられる遺伝性疾患であり、様々な臓器への影響がみられます。
シスチノシンは、細胞がタンパク質をリサイクルし、新しいタンパク質の合成のために分解されたアミノ酸を再利用する過程で極めて重要な役割を果たす特殊な輸送タンパク質です。このトランスポーターはリソソームから細胞内へと必須アミノ酸であるシステインを運び出します。システインは、シスチンとしてリソソーム内に蓄積されることがありますが、シスチノシンによって適切に細胞外へ運ばれることが重要です。
シスチノシンは、リソソーム内部でシスチンを取り込み、外部へ放出する際に異なる構造(コンフォメーション)を取るタンパク質です。リソソームの内側に向いているとき、シスチノシンはシスチンを装填するための形状をしています。一方で、外側に開いているときには、装填したシスチンを外部へ放出するための形状に変化します。この機能により、シスチノシンはリソソーム内のシスチン濃度を調節し、細胞内のアミノ酸バランスの維持に寄与します。
シスチノシンの機能に異常が生じると、シスチンがリソソーム内に過剰に蓄積し、これがシスチノーシスという遺伝性の病態を引き起こします。シスチンの蓄積は細胞に毒性を示し、腎臓、眼、筋肉、神経系などの組織や臓器に広範囲にわたる損傷を与えることがあります。これにより、腎不全や筋肉の衰弱などの深刻な健康問題が発生する可能性があります。シスチノーシスの患者は、シスチンの蓄積を管理し、その影響を最小限に抑えるために特定の治療を必要とすることがあります。
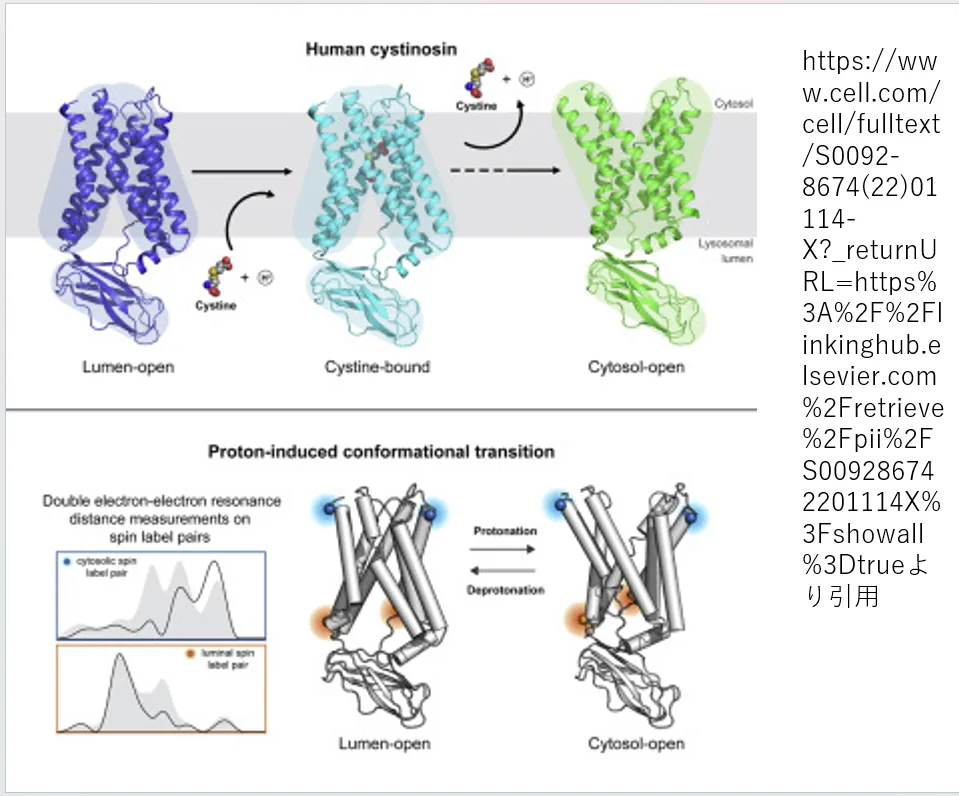
https://www.cell.com/cell/fulltext/S0092-8674(22)01114-X?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS009286742201114X%3Fshowall%3Dtrue
より引用
遺伝子と関係のある疾患
遺伝子の発現とクローニング
CTNS遺伝子の発現は組織特異的で、特に膵臓、腎臓(成人及び胎児のもの)、骨格筋で強く発現していることが分かりました。胎盤と心臓では発現量が少なく、肺と肝臓ではさらに弱い発現が確認されました。この研究は、シスチンの輸送機構の理解を深めると共に、腎症性シスチン症の治療に向けた新たな知見を提供するものです。
遺伝子の構造
さらに、CTNSのプロモーター領域は、隣接する遺伝子CARKL(SHPK)のプロモーター領域と41ヌクレオチドを共有しています。CARKL遺伝子はCTNS遺伝子とは反対方向のDNA鎖上に位置しており、その開始部位はCTNSの開始部位から501bp離れています。このような遺伝子の配置は、両遺伝子の転写調節において相互作用がある可能性を示唆しています。
CTNS遺伝子の構造とプロモーター領域の詳細な解析は、シスチノーシスの理解と治療法の開発において重要です。特に、Sp-1制御エレメントとの相互作用は、遺伝子の転写活性を調節する新たな治療標的となる可能性があります。また、隣接するCARKL遺伝子との関係性も、両遺伝子の機能的相互作用に関する今後の研究の基礎を提供します。
マッピング
腎症性シスチン症は、シスチンが腎臓の尿細管に蓄積し、シスチン結石を形成する遺伝性疾患です。この疾患の患者では、シスチンの再吸収がうまく行われず、結石形成に至ります。CTNS遺伝子の特定は、この疾患の分子生物学的基盤の理解を深め、将来的には治療法の開発に繋がる可能性があります。Townらの研究は、遺伝子マッピングと遺伝子同定の分野における重要な貢献を示しています。
分子遺伝学
Townらによる研究では、CTNS遺伝子の11種類の変異が同定され、最も一般的な変異は65kbの欠失であり、検体の33%で見られました。この変異はタンパク質の機能喪失を引き起こすと予測されています。
Shotelersukらの研究では、アメリカに基づく腎症性嚢胞性疾患患者108人の分析から、多様な変異が同定され、その中でも「ヨーロッパ型」の65kb欠失が44%の患者で見られることが示されました。さらに、疾患の臨床的重症度と変異の位置や種類との関連が示唆されています。
Attardらによる研究では、乳児腎症性シスチン症患者や遅発性シスチノーシス患者を含む様々なシスチノーシス患者のCTNS遺伝子をスクリーニングし、23種類の変異、そのうち14種類が新規変異として同定されました。乳児腎症性シスチン症患者の多くでは、機能的蛋白質の欠損を引き起こす重度の変異が見られましたが、遅発性患者では、機能的に重要でない領域に影響を与える変異が見られ、より軽症の表現型につながることが示されています。
CTNS遺伝子の同定とその周辺領域の研究は、シスチノーシスの遺伝的基盤についての重要な知見を提供しています。シスチノーシスは、細胞内のシスチン輸送を調節するリソソーム膜タンパク質であるシスチノシンをコードするCTNS遺伝子の変異によって引き起こされる遺伝性疾患です。Forestierら(1999)とTouchmanら(2000)、およびPhornphutkulら(2001)の研究は、この疾患に関連するさまざまな遺伝子変異の特徴と、それらがどのようにして疾患の表現型に影響を与える可能性があるかを明らかにしました。
Forestierらの研究では、シスチン症患者の約3分の1にホモ接合状態で見られる約65kbの欠失と、1家族に見られる9.5〜16kbの小さな欠失が特定されました。これらの欠失はCTNS遺伝子の5-プライム末端にまたがり、大きい欠失はエクソン1〜10を、小さい欠失はエクソン1〜3をカバーしています。これらの欠失は非相同組換えの結果であることが示され、約65kbの欠失は創始者効果によるものであり、おそらく最初の千年紀の中頃に生じたものであることが示されました。
Touchmanらの研究では、CTNS遺伝子周辺200kbの塩基配列が決定され、一般的なシスチン症の欠失は約57kbであることが明らかにされました。この欠失にはCTNS遺伝子だけでなく、CARKL(現在はSHPKとして知られる)も含まれていることが示され、これが表現型の多様性に影響を与える可能性が示唆されました。
Phornphutkulらの研究では、シスチン症患者のCTNSプロモーター領域に変異が同定され、これらの変異がプロモーター活性を劇的に低下させることが示されました。これらのプロモーター変異は、CARKL遺伝子のプロモーター活性には影響を与えませんでした。
Gahlら(2002年)によると、シスチン症には50以上の異なるCTNS突然変異があり、最も一般的な変異は57,257bpの欠失で、北ヨーロッパ系の患者の約50%で見られることが報告されています。この変異は、シスチン症の患者における精神発達障害の重症度と関連していることが示されています。
中等度シスチン症や眼性シスチン症の患者は、重度と軽度のCTNS変異の両方を持つことが多く、これによりシスチノシンの輸送機能の一部が保持されると述べられています。台湾の2人の兄弟に見られたN323K変異のように、保存されたアミノ酸の変異がホモ接合体で見つかる例もあります。
Kalatzisら(2002年、2004年)の研究は、シスチン症に関連する新たな変異を同定し、これらの変異がシスチノシンのリソソーム局在や輸送活性にどのように影響するかを解析しました。彼らの研究は、輸送障害が病原性の最も一般的な原因であることを示し、特に小児型シスチン症は活性の完全な喪失によって特徴付けられることを明らかにしました。
Macias-Vidalら(2009年)の研究は、スペインとモロッコのシスチン症患者におけるCTNS遺伝子の変異スペクトルを明らかにし、特に小児型シスチン症において切断変異や重要な膜貫通領域に影響を及ぼす変異が一般的であることを示しました。
これらの研究は、シスチン症の分子的および臨床的理解を深め、遺伝子診断や将来の治療戦略の開発に重要な情報を提供しています。特に、CTNS遺伝子の変異の同定と解析は、シスチン症の病態生理の理解を進める上で不可欠です。
遺伝子型と表現型の相関
Goodmanら(2021)は、異なるアプローチを用いてシスチン症の治療可能性を探りました。彼らは、Shpk遺伝子欠損マウスから採取した造血幹細胞および前駆細胞(HSPCs)をCtns欠損マウスに移植しました。治療を受けたCtns欠損マウスでは、未治療のマウスと比較して複数の組織でシスチン負荷が有意に減少し、Ctnsの発現が回復しました。さらに、腎臓の形態学的構造が改善されました。Goodmanらは、SHPKの欠失がHSPCsのシスチン症に対する回復能力を変化させないこと、およびシスチン症患者におけるex vivo遺伝子治療アプローチの有益性を示唆しました。
これらの研究は、シスチン症の治療法開発に向けた基礎研究として重要な貢献をしており、遺伝子治療が将来的な治療オプションとして有望であることを示しています。
アレリックバリアント
.0001 シスチン症、腎症性
CTNS, GLY95TER
パキスタンの2血族において、Townら(1998)は腎症性シスチン症(219800)がCTNS遺伝子のgly95-to-ter(G95X)変異と関連していることを発見した。4cMの区間をカバーする5つのマイクロサテライトの解析から、共通のハプロタイプが示され、これらの家系が関連している可能性が示唆された。変異はヌクレオチド622におけるGからTへの転位であり、GGA(gly)からTGA(stop)への変換であった。
.0002 シスチン症、腎症性
CTNS、2bp欠損、397TG
Townら(1998)は、フランス中部の2家系において、腎症性シスチン症(219800)がCTNS遺伝子の同じ2-bp欠失の半接合性と関連していることを発見した:397/399のTGの欠失により、変異部位に停止コドンが生じた。この2家系は欠失と分離する共通のハプロタイプを共有していた。第2対立遺伝子の欠失は各家族の罹患者に認められた。
.0003 シスチン症,腎症性
CTNS、TRP138TER
Townら(1998)は、北アイルランドの1家族とEireの1家族に、腎症性シスチン症(219800)の基礎となる変異と同じ変異、すなわち、ヌクレオチド753におけるTGGからTGAへの転移により、trp138からter(W138X)へのナンセンス変異があることを発見した。
McGowan-Jordanら(1999)は、W138X変異を21/40のフランス系カナダ人シスチン症染色体で発見した。すべての症例で、変異は特徴的なハプロタイプ上にあった。彼らはこの変異を持つアイルランド人2家族にも同じハプロタイプを見つけ、ケルト染色体がフランス系カナダにおけるシスチン症染色体の広範な部分を占めているという仮説を支持した。この研究はまた、フランス系カナダ人の集団において、しばしば認識されない非金属由来の寄与も示唆している。
.0004 シスチン症, 腎症性
遅発性若年性または青年性腎症型シスチノーシス、含む
CTNS、4-BP欠損、18ガクト
Townら(1998)は、3つの異なる大陸からの4家族において、腎症性シスチン症(219800)がCTNS遺伝子のヌクレオチド357における4ヌクレオチド、GACTの欠失と関連していることを発見した。これはフレームシフトと早期終結をもたらした。4家族は共通のハプロタイプを共有しておらず、再発性の突然変異であることが示された。Macias-Vidalら(2009)は、ATG開始コドンからの番号付けに基づき、欠失はヌクレオチド18で起こると指摘している。
若年性腎症性シスチン症のスペイン人患者(219900)において、Macias-Vidalら(2009)は、CTNS遺伝子の416C-T転移の複合ヘテロ接合性を同定し、その結果、ser139-to-phe(S139F; 606272.0018)置換と4bp欠失が生じた。
.0005 シスチン症、腎症性
遅発性若年性または青年期腎症型シスチノーシス、以下を含む
眼性非ネフローゼ型シスチン症、含まれる
CTNS、57-KB欠失
この腎症性シスチン症によくみられる変異(219800)は、当初65kbの欠失として報告された。Touchmanら(2000)はCTNS遺伝子の周囲200kbの塩基配列を決定し、欠失は65kbではなく約57kbであることを発見した。著者らはこの欠失領域内にCARKLと命名したSHPK (605060)を同定した。この所見から、57kbの欠失にはCTNSに加えてCARKLの欠失も含まれていることが示され、これが患者における表現型のばらつきを説明している可能性がある。
フランスとイギリスの報告(Town et al., 1998)では、70人の腎症性シスチン症患者のうち23人(33%)がCTNS遺伝子に65kbの欠失を有していた。Shotelersukら(1998)が調査したアメリカを拠点とする患者では、108人中48人(44%)が65kbの「ヨーロッパ型」欠失をホモ接合体で有していた。これらの患者から得られた96の対立遺伝子のうち、82の対立遺伝子に由来国が割り当てられた;38(46%)はドイツに由来し、28(34%)はイギリス諸島に由来した。ホモ接合体欠失を持つ明らかに無関係と思われる2人の患者はアイスランド出身であった。65kb欠失のホモ接合体48例に加え、多くの患者は欠失の単一コピーを持っている可能性がある。欠失に対するPCRベースのヘテロ接合性検査を開発する前に、上流の欠失ブレークポイントを決定する必要があった。
Gahlら(2002)は、57kbの欠失は、北ヨーロッパ系のシスチン症患者の約50%にホモ接合状態で認められると述べている。CTNSの最初の10エクソンを除去し、タンパク質の発現を消失させるこの創始者変異は、明らかに西暦約500年にドイツで発生し(Shotelersukら、1998)、アイスランドを含む他の地域への移動によって広がった。
Bendavidら(2004)は、細胞遺伝学研究所で57kbの欠失を検査できるFISH法について述べており、この欠失は米国および北ヨーロッパのシスチン症患者の約60%に認められる。
Wamelinkら(2008年)は、57-kb欠失のホモ接合体のシスチン症患者では、他のCTNS変異を有する患者と比較して、尿中のセドヘプツロースとエリスリトールが増加していることを見いだした。培養線維芽細胞を用いた酵素研究では、セドヘプツロースリン酸化活性が、他の変異を持つシスチン症患者や対照群と比較して80%低下していることが明らかになった。この結果から、CARKL遺伝子はペントースリン酸経路で機能するセドヘプツロキナーゼをコードしていることが示された。
Buntinxら(2016)は、北ヨーロッパのシスチン症患者に最も多い変異はCTNS遺伝子の57kb欠失であると指摘した。この欠失は、カプサイシンおよび熱感受性イオンチャネルをコードするTRPV1遺伝子(602076)の開始コドン上流の非コード領域に及んでいる。Buntinxら(2016)は、欠失のヘテロ接合体患者は正常な感覚応答を示したが、変異のホモ接合体患者はカプサイシンで誘発される血管拡張と疼痛が60%減少し、熱検出閾値も上昇することを見いだした。寒冷、機械的刺激、TRPA1 (604775)アゴニストであるシンナムアルデヒドに対する反応は変化しなかった。Buntinxら(2016)は、57-kb CTNS欠失のホモ接合体であるシスチン症患者はTRPV1の機能が強く低下しており、おそらくこれらの患者における感覚の変化と体温調節障害を説明しているのだろうと結論している。
57kb欠失の複合ヘテロ接合体
38歳で羞明を呈し、慢性的な光線過敏症を患っていた38歳の女性(219750)において、Aniksterら(2000)は、57-kb欠失の複合ヘテロ接合と、コドン197でグリシンからアルギニンへの置換を生じる928G-A転移を同定した(G197R; 606272.0011)。複合ヘテロ接合は、同じ家系の眼性シスチン症の患者2人にも認められた。
若年発症の腎症性シスチン症のスペイン人患者(219900)において、Macias-Vidalら(2009)は、CTNS遺伝子の416C-T転移の複合ヘテロ接合性を同定し、その結果、ser139からpheへの置換(S139F; 606272.0018)と57kbの欠失が生じた。
.0006 シスチン症、腎症性
CTNS, GLY169ASP
Shotelersukら(1998)が腎症性シスチン症患者(219800)で発見したCTNS遺伝子の7つのミスセンス変異の一つは、gly169からaspへの(G169D)アミノ酸置換であった。有意なCTNS発現はこの変異のホモ接合性と関連していた。7つのミスセンス変異はすべて、膜貫通ドメイン内部または膜貫通ドメインの前の最初のアミノ酸にアミノ酸変化を生じた。
.0007 非定型腎症性シスチン症
CTNS、VAL42ILE
Attardら(1999)は、3人の患者において、シスチノーシス(219800参照)の非典型的な発現を発見した。発症は7歳未満であったが、経過は非典型的に軽度であった。患者3人のうち1人(L18)はCTNS遺伝子のGからAへの転移がホモ接合体であり、その結果、N末端の非保存領域にval42からile(V42I)への置換が生じた。タンパク質のこの部分はリソソームの内腔にあると予測され、変異は潜在的なN-グリコシル化部位に隣接していたが、影響はなかった。従って、変異の位置はより軽い表現型と一致していた。
.0008 後期発症若年性または青年性腎症型シスチン症
CTNS, IVS7AS, C-G, -10
後期発症の典型的なシスチン症患者(219900)において、Attardら(1999)はCTNS遺伝子のイントロン変異、ヌクレオチド801-10におけるCからGへの転座を同定した。この結果、正常部位の上流に代替スプライス部位が形成された。cDNAのエクソン7-8の境界を横切る塩基配列を決定したところ、9塩基がホモ接合性で挿入され、第2膜貫通ドメインのすぐ隣の位置に3アミノ酸(pro-cys-ser)が付加されることがわかった。これは保存された領域で起こったが、最初の細胞質ドメインは非常に小さい(通常は4アミノ酸)ので、機能的には重要ではないかもしれない。この時点でプロリンのイミノ基が付加されると、ポリペプチド鎖のフォールディングが破壊されることが予想されるが、膜貫通ドメインに入り込んだり干渉したりすることはなさそうである。
.0009 眼性非ネフローゼ性シスチン症
CTNS、IVS10AS、C-G、-3
眼性非ネフローゼ性シスチン症(219750)の26歳男性において、Aniksterら(2000)はCTNS遺伝子のIVS10のアクセプタースプライス部位の-3位におけるC-G転移を報告した。この変異はヌクレオチド545のTCCTT欠失との複合ヘテロ接合で発見された。
.0010 シスチン症、腎症性
CTNS、5bp欠失、NT545
Shotelersukら(1998)は、古典的なシスチン症患者(219800)において、CTNS遺伝子の73位にI69Rアミノ酸置換と停止コドンをもたらすヌクレオチド545で始まる5-bp欠失を同定した。
.0011 眼非ネフローゼ性シスチン症
CTNS、Gly197Arg
38歳で羞明を呈したが、慢性光線過敏症を患っていた38歳の女性(219750)において、Aniksterら(2000)は、ヌクレオチド928でGからAへの転移を同定し、その結果、コドン197でグリシンからアルギニンへの置換が生じた(G197R)。この患者は57kbの欠失の複合ヘテロ接合体であった(606272.0005)。この変異は同じ家系の眼性シスチン症の患者2人にも認められた。両患者ともG197R変異と57kb欠失の複合ヘテロ接合体であった。
.0012 シスチン症、腎症性
CTNS、-295G-C、プロモーター
Phornphutkulら(2001)は、腎症性シスチン症(219800)のスコットランド系アイルランド人とプエルトリコ人の女性において、CTNS遺伝子の57kb欠失(606272.0005)とプロモーター変異(Sp-1調節エレメントに関与するヌクレオチド-295のG-to-C変化)のヘテロ接合を同定した。後者の変異は、-348から+1のCTNS-CAT構築物を作製してプロモーター活性への影響を試験したところ、HeLa細胞にトランスフェクトした場合、野生型CAT活性の19%を産生することが判明した。
.0013 シスチン症、眼性非ネフトロパシー性
CTNS、-303G-T、プロモーター
ドイツ/ノルウェー人の血を引く眼性シスチン症の女児(219750)において、Phornphutkulら(2001)はCTNS遺伝子のG197R変異(606272.0011)とプロモーター変異(ヌクレオチド-303におけるGからTへのトランスバージョン)のヘテロ接合を同定した。後者の変異は、-348から+1のCTNS-CAT構築物を作製してプロモーター活性への影響を試験したところ、HeLa細胞にトランスフェクトした場合、野生型CAT活性の5%を産生することが判明した。
.0014 シスチン症、眼性非ネフトロパシー性
CTNS、1-bp ins、-303t、プロモーター
Aniksterら(2000)によって報告された眼性シスチン症(219750)の女性において、Phornphutkulら(2001)はCTNS遺伝子のG197R変異(606272.0011)とプロモーター変異、-303位の後のTの挿入のヘテロ接合性を同定した。後者の変異は、-348から+1のCTNS-CAT構築物を作製することにより、プロモーター活性への影響を試験したところ、HeLa細胞にトランスフェクトした場合、野生型CAT活性の16%を産生することが判明した。
.0015 シスチン症、腎症性
CTNS、Gly339ARG
オハイオ州南西部のオールド・オーダー・アーミッシュ集団の4人の腎症性シスチン症(219800)の子供において、Ruparら(2001)はヌクレオチド1354でGからAへの転移を同定した。この転移は残基339のグリシンからアルギニンへの置換(G339R)をもたらした。罹患児ではホモ接合型で、罹患していない兄姉ではヘテロ接合型で発見された。
.0016 シスチノーシス、遅発性若年性または青年性腎症型
CTNS, ASN323LYS
Thoeneら(1999)は、血漿クレアチニン濃度がそれぞれ1.2mg/デシリットルと3.3mg/デシリットルになった13歳と14歳まで、直線的な成長と体重増加が民族の平均の2標準偏差以内であった、中間のシスチノーシス(219900)を持つ台湾の2人の兄弟について記述している。彼らはCTNS遺伝子の1308C-G変異のホモ接合体であり、その結果、323位の保存されたアスパラギン(N323K)がリジンに置換されていた。おそらく、この変異によりシスチンの輸送がある程度可能になり、軽度の臨床症状が認められたものと思われる。
.0017 非定型腎症性シスチン症
CTNS、Gly110VAL
非典型的な腎症性シスチン症(219800参照)で、ファンコニー症候群(134600)と末期腎臓病を呈するが、驚くべきことに晩年になっても腎外症状を呈さない患者において、Kalatzisら(2002)はCTNS遺伝子のN末端領域に位置するgly110-to-val(G110V)変異を検出した。この置換は非保存残基に影響するため、有意な影響はないと予想されたが、エクソン6の最後のヌクレオチドに関与し、CTNSのスプライシングに影響を与えた。アミノ酸位置111でフレームシフトを引き起こし、7番目の膜貫通セグメントの手前でタンパク質が早期に終結する異常な転写産物が産生され、正しくスプライシングされたCTNS転写産物は検出されなかった。しかし、白血球RNAのみが研究され、G110Vがすべての組織で同じスプライシングイベントを引き起こさない可能性が考えられた。腎臓でミスプライシングされた転写物は腎臓の重篤な表現型を説明するかもしれないが、他の臓器では少量でも正しくスプライシングされた形が存在すれば腎外障害がないことを説明できるかもしれない。
.0018 シスチノーシス、遅発性若年性または青年性腎症型
嚢胞性腎症、非定型、含む
ctns, ser139phe
Macias-Vidalら(2009)は、血縁関係のない若年性腎症性シスチン症(219900)のスペイン人患者2人において、CTNS遺伝子の416C-T転移の複合ヘテロ接合性を同定し、その結果、ser139-to-phe(S139F)置換、4bp欠失(606272.0004)および57kb欠失(606272.0005)がそれぞれ生じた。S139F変異は以前、Attardら(1999)によって、7歳以前に発症するが、小児腎症型よりも疾患の経過が軽い「非古典的」シスチノーシス(219800を参照)の患者において同定されており、この変異は機能的タンパク質の産生を可能にするか、機能的に重要でないシスチノシンの領域に位置している可能性が示唆されていた。
参考文献
https://www.omim.org/entry/606272







