目次

ATP1A2遺伝子は、脳・心臓・骨格筋・血管平滑筋でナトリウムカリウムポンプα2サブユニットをつくる、生体のイオンバランスと電気信号の伝達を支える重要遺伝子です。変異により家族性片麻痺性片頭痛2型・小児交互性片麻痺・てんかん性脳症・周期性四肢麻痺・心機能障害など、多彩な神経・筋・心疾患を発症します。
Q. ATP1A2遺伝子とはどのような遺伝子ですか?まず結論だけ知りたいです
A. ATP1A2は1番染色体(1q23.2)に存在し、ナトリウムカリウムポンプ(Na+/K+-ATPase)のα2サブユニットをつくる遺伝子です。脳のアストロサイト、心臓、骨格筋、血管平滑筋に多く発現し、細胞内外のイオンバランスと電気的興奮性の制御を担います。変異により、家族性片麻痺性片頭痛2型・小児交互性片麻痺・重度てんかん性脳症・周期性四肢麻痺・心機能障害など、極めて幅広い神経・筋・心疾患を発症します。
- ➤遺伝子の基本情報 → 1q23.2、Na+/K+-ATPase α2サブユニット、P型ATPase
- ➤主な発現組織 → 脳のアストロサイト・心筋・骨格筋・血管平滑筋・肺・脂肪細胞
- ➤病態メカニズム → ハプロ不全・構造的機能不全・異常な内向きカチオンリーク電流
- ➤関連疾患 → FHM2・AHC・DEE98・HypoPP・心不全・FARIMPDなど
- ➤最新治療 → メマンチン、経口ATP療法、CRISPR塩基編集による遺伝子治療
1. ATP1A2遺伝子とは:基本情報と発見の歴史
ATP1A2遺伝子(正式名称:ATPase Na+/K+ Transporting Subunit Alpha 2)は、ヒトの1番染色体長腕の23.2領域(1q23.2)に存在する遺伝子です。私たちの体の細胞には、細胞膜を挟んで内側と外側でナトリウム(Na+)とカリウム(K+)の濃度バランスを精密に保つ「ポンプ」が備わっています。このポンプを「ナトリウムカリウムポンプ(Na+/K+-ATPase)」と呼び、ATP1A2はそのポンプを構成する4種類のαサブユニットのうち、α2と呼ばれる触媒サブユニットをつくる設計図です。
💡 用語解説:ナトリウムカリウムポンプ(Na+/K+-ATPase)
細胞膜にある「タンパク質の機械」のひとつです。ATPというエネルギー分子1個を使うたびに、Na+を3個外へ汲み出し、K+を2個内へ取り込みます。この働きによって細胞は安静時に決まった電位(静止膜電位)を保つことができ、神経が信号を伝えたり、筋肉が収縮したりする土台となります。発見者の名前から「Post-Albersサイクル」と呼ばれる反応の流れに沿って動きます。
ATP1A2遺伝子変異と病気との関連は、長らく家族性片麻痺性片頭痛2型(FHM2)の原因として知られてきました。しかし2010年代以降、次世代シーケンス技術の普及と国際的な患者データ統合により、この遺伝子の関連疾患スペクトラムは劇的に拡大しています。現在では、小児交互性片麻痺(AHC)、重篤な発達性およびてんかん性脳症(DEE98)、低カリウム血症性周期性四肢麻痺(HypoPP)、心不全や心停止を含む自律神経・心血管系障害まで、脳から心臓・筋肉まで全身に波及する多臓器疾患の原因遺伝子として認識されています。
🔍 ATP1A2の基本データ 遺伝子座:1q23.2/コードするタンパク質:Na+/K+-ATPase α2サブユニット/タンパク質ファミリー:P型カチオン輸送ATPase/主な発現組織:脳(アストロサイト)、心筋、骨格筋、血管平滑筋、肺、脂肪細胞
2. ATP1A2の働き:Na+/K+ポンプα2サブユニットがつくる電気の土台
ATP1A2がつくるα2サブユニットは、Na+/K+-ATPaseの4種類のαアイソフォーム(α1〜α4)のひとつです。それぞれのアイソフォームは発現している組織や役割が異なり、α2には「活動電位が起こったときに強力に働く予備電源」のような独自の特性があります。
4種類のαサブユニットの違い
| アイソフォーム | 遺伝子 | 主な発現組織 | 特徴 |
|---|---|---|---|
| α1 | ATP1A1 | ほぼ全細胞 | 基本的なイオン恒常性。α2欠損時の代償も担う |
| α2 | ATP1A2 | 心筋・骨格筋・血管平滑筋・脳(アストロサイト)・肺 | 活動電位時の急激なイオン変動を緩衝する予備電源 |
| α3 | ATP1A3 | 神経細胞(ニューロン) | ニューロン主要型。AHCや急速発症型ジストニア・パーキンソニズムの原因 |
| α4 | ATP1A4 | 精子 | 精子の運動性に必要 |
💡 用語解説:活動電位(かつどうでんい)
神経や筋肉の細胞が情報を伝えるときに発する「電気のパルス」です。Na+やK+のイオンが細胞膜を一気に通過することで生まれます。1回の活動電位が終わるたびに、細胞は乱れたイオンバランスをすばやく元に戻さなければなりません。α2サブユニットはこの「戻し作業」の主役であり、心拍ごとに収縮する心筋や、高頻度で発火する神経細胞では特に欠かせない存在です。
α2サブユニットの最大の特徴は、電圧依存性が非常に急峻であることです。普段の静止状態ではほぼ動かず、細胞が脱分極(電気的に活発になる状態)したときだけ強力に作動します。心筋や骨格筋が収縮するとき、神経細胞が連続発火するとき——そういった「ピーク需要」の瞬間に集中して働く、いわば予備電源としてのポンプなのです。
3. アストロサイトでの中心的役割:脳のグルタミン酸クリアランス
ATP1A2が脳でどう働いているかを理解することは、片麻痺性片頭痛やてんかん性脳症のメカニズムを知るうえで決定的に重要です。意外に思われるかもしれませんが、ATP1A2は神経細胞(ニューロン)そのものよりも、ニューロンを取り囲む「アストロサイト」というグリア細胞に高密度に発現しています。
💡 用語解説:アストロサイト
脳と脊髄に多く存在する星形の細胞で、グリア細胞と呼ばれる「神経のサポート役」の代表選手です。ニューロンに栄養を届け、神経細胞間の隙間(シナプス間隙)に放出された神経伝達物質を回収し、脳のイオンバランスを保ちます。長らく「縁の下の力持ち」と見られていましたが、近年は脳機能の能動的な調節役として注目されています。
脳のシナプス(神経細胞どうしのつなぎ目)では、興奮性の神経伝達物質であるグルタミン酸が放出されて信号が伝わります。しかしグルタミン酸はシナプス間隙にとどまり続けると、後ろのニューロンを過剰に興奮させてしまうため、すぐに回収されなければなりません。この回収作業を担うのが、アストロサイトの表面にあるEAAT(興奮性アミノ酸トランスポーター)と呼ばれる「掃除機」です。
EAATがグルタミン酸を吸い込むためには、アストロサイト内外で大きなNa+の電気化学的勾配(濃度差)が必要で、そのNa+勾配を作り出しているのがATP1A2でつくられるα2ポンプです。つまり、ATP1A2の働きが弱まると、Na+勾配が崩れ、EAATがグルタミン酸を回収できなくなり、シナプスに過剰なグルタミン酸が滞留する——という連鎖が起こります。
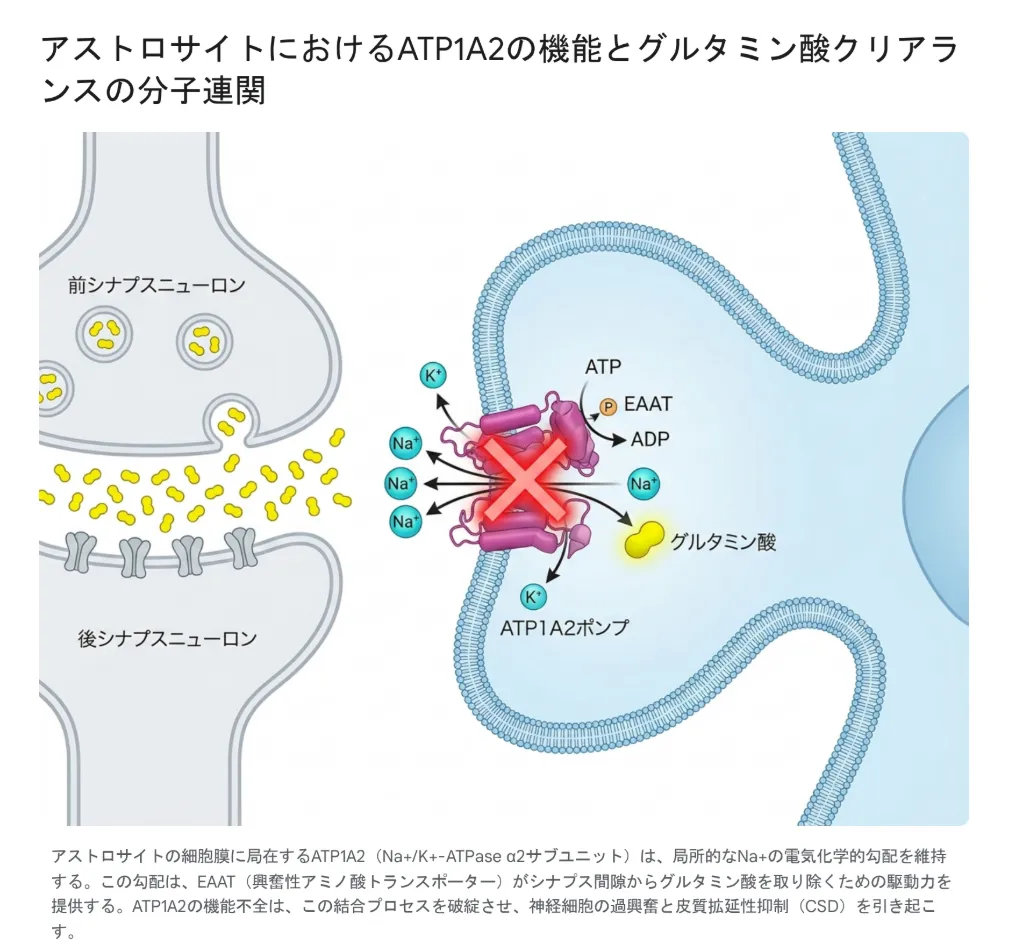
アストロサイト細胞膜のATP1A2が局所のNa+電気化学的勾配を維持し、EAATがシナプス間隙からグルタミン酸を回収する駆動力を提供する。ATP1A2の機能不全はこの一連のプロセスを破綻させ、神経の過興奮と皮質拡延性抑制(CSD)を引き起こす。
💡 用語解説:皮質拡延性抑制(CSD:Cortical Spreading Depression)
大脳皮質の表面を、神経細胞の異常な興奮の波がゆっくり広がっていく現象(毎分2〜5mm程度)です。波が通った後は逆に神経活動が一時的に抑制されます。片頭痛の前兆(視覚のチカチカ・手足のしびれなど)の正体と考えられており、ATP1A2変異によるグルタミン酸の滞留はこのCSDの発生しやすさを高めると考えられています。これがFHM2の中心的な病態仮説(「グルタミン酸仮説」)です。
4. 変異が引き起こす3つの病態メカニズム
ATP1A2遺伝子に生じる病原性バリアント(病気の原因となる変異)の大多数は、DNAの1塩基が変化して別のアミノ酸に置き換わるミスセンス変異です。しかし、同じミスセンス変異でも、タンパク質のどこが影響を受けるかによって細胞内で起こる「異常の質」がまったく異なります。これがATP1A2変異患者の臨床症状の幅広さを生む根本的な理由です。
💡 用語解説:ミスセンス変異と新生突然変異(de novo変異)
ミスセンス変異とは、DNAの1文字が変わることでタンパク質を構成するアミノ酸が別の種類に置き換わる変異です。タンパク質の形がわずかに変わり、機能に影響します。
新生突然変異(de novo変異)とは、両親には存在せず、お子さんで初めて新しく生まれた変異のこと。ATP1A2の重症型疾患の多くはこの新生突然変異で発症します。
メカニズム①:ハプロ不全(タンパク質の量が足りなくなる)
FHM2の多くの変異では、変異側の遺伝子から正常なタンパク質がつくられなくなるか、つくられても活性が著しく低下します。私たちは父母から1コピーずつ遺伝子を受け継いでいるので、片側が機能しないと細胞内のα2サブユニットの総量は半分ほどになります。これをハプロ不全と呼びます。激しい筋収縮や高頻度発火の場面で「予備電源」が足りなくなり、イオンバランスの回復が遅れて症状が出ます。
メカニズム②:構造的機能不全(イオン結合サイトの直接的破壊)
DEE98のような重症のてんかん性脳症や知的障害を伴う症例では、変異がポンプ機能の中枢であるイオン結合サイトやイオン通過経路に直接ダメージを与えていることが分子モデリング解析で示されています(例:p.Ile293Met、p.Glu1000Lys、p.Met813Lysなど)。単なる量的不足ではなく、ポンプそのものの動作原理が壊れるため、純粋な片頭痛にとどまらず重度の発達遅滞や難治性てんかんを呈します。
メカニズム③:異常な内向きカチオンリーク電流(最近発見された新しい病態)
2018年以降に解明された極めて画期的な知見が、低カリウム血症性周期性四肢麻痺(HypoPP)の患者から見つかった新生突然変異による「内向きカチオンリーク電流」です。正常なポンプは外向きにNa+を汲み出す方向にしか働きませんが、これらの変異ポンプは低カリウム条件下で逆に細胞内に陽イオンを漏らしてしまうのです。骨格筋細胞は異常に脱分極し、一過性の麻痺が起こります。
📌 まとめ ATP1A2変異の臨床的重症度は、ハプロ不全(量的不足)→構造的機能不全(質的破壊)→リーク電流(誤動作)と、メカニズムの種類によって大きく異なります。患者ごとの変異の質を分子レベルで読み解くことが、診断と予後予測の両方に直結します。
5. ATP1A2変異による関連疾患のスペクトラム
ATP1A2変異は、もはや「片麻痺性片頭痛だけの遺伝子」ではありません。31名のATP1A2/ATP1A3変異患者を対象とした大規模ケースシリーズでは、てんかん発作が77%、発達遅滞が65%、運動障害が35%、片頭痛が23%に認められ、致死的な心停止(6%)や無呼吸エピソード(6%)も報告されています。古典的な「片頭痛」イメージとは大きく異なる、連続的な表現型スペクトラムが浮かび上がっています。
💢 家族性片麻痺性片頭痛2型(FHM2)
ATP1A2変異の代表的疾患。常染色体顕性(優性)遺伝で、視覚異常・しびれ・言語障害の前兆に続き、片側の脱力(片麻痺)を伴う激しい頭痛発作が起こります。発症は10〜20代が多く、年齢とともに頻度が減る傾向があります。▶ FHM2の詳細
🧒 小児交互性片麻痺(AHC)
生後18ヶ月以内に発症する希少な神経発達障害。左右が交互に麻痺する反復エピソード、ジストニア、眼振、自律神経症状、進行性の認知機能低下を特徴とします。ATP1A3変異が主原因ですが、ATP1A2変異例も近年報告が増えています。▶ AHCの詳細
⚡ 発達性てんかん性脳症(DEE98)
最重症型。生後数日から2歳までに発症し、難治性てんかん・反復するてんかん重積状態・重度発達遅滞を呈します。脳MRIでは広範な皮質浮腫や神経細胞ダメージを示す所見が認められることがあります。▶ DEE98の詳細
💪 低カリウム血症性周期性四肢麻痺(HypoPP)
骨格筋の一過性の脱力・麻痺が、低カリウム条件で繰り返し起こります。従来CACNA1SやSCN4Aが原因と考えられてきましたが、これらに変異がない症例でATP1A2新生変異が見つかり、新たな原因遺伝子として確立されました。▶ 周期性四肢麻痺NGSパネル
心血管系への影響:見落とされやすい合併症
心臓におけるα2アイソフォームの割合は全Na+/K+-ATPaseの10〜15%程度と少数ですが、循環動態の調節には欠かせません。FHM2変異マウス(G301R)の研究では、生後3ヶ月では正常な心機能を示すものの、生後8ヶ月で左室拡張・駆出率低下・夜間血圧低下といった心不全様の変化が確認されました。背景にはα2量低下→活性酸素種(ROS)増加→Src/Ras/Erk1/2経路の異常活性化→ミトコンドリア障害という分子カスケードがあることが、プロテオミクス・メタボロミクス解析で明らかになっています。31名のケースシリーズで報告された心停止(6%)や無呼吸エピソード(6%)も、自律神経の不均衡や心筋への直接的なチャネル異常が関与している可能性があります。
劣性ホモ接合体による別の表現型:FARIMPD
ここまで紹介してきたFHM2やAHC、DEE98、HypoPPはいずれもヘテロ接合体(片方のアレルの変異)による常染色体顕性(優性)遺伝の疾患です。これとは別に、両アレルに機能喪失型の変異があるホモ接合体では、胎児致死性のFARIMPD(胎児無動・呼吸不全・小頭症・多小脳回・顔面形成異常)という別の症候群が起こることが2019〜2020年に報告されました。▶ FARIMPDの詳細
💡 用語解説:常染色体顕性(優性)遺伝と常染色体劣性(潜性)遺伝
常染色体顕性(優性)遺伝とは、2本ある常染色体のうち片方に変異があるだけで症状が出るタイプ。FHM2やAHCなどATP1A2の大多数の疾患はこちらです。
常染色体劣性(潜性)遺伝とは、両親から1つずつ変異を受け継いで両方の遺伝子に変異がある場合に発症するタイプ。ATP1A2のFARIMPDはこちらに該当します。

6. ATP1A2遺伝子検査:当院でご提供している検査メニュー
ATP1A2は、症状の幅広さから「単独で検査」されるよりも、関連症状の遺伝子群をまとめて解析するNGS(次世代シーケンス)パネル検査に含めて評価する方が、診断率も鑑別の効率も高まります。当院では、ATP1A2を含む4つの検査プランをご提供しています。
⚡ 思春期・成人発症てんかんNGSパネル(84遺伝子)
ATP1A2を含む、思春期以降に発症するてんかんの原因遺伝子84種を網羅。前兆・部分発作・全般発作のいずれの病型でも評価可能です。
💪 周期性四肢麻痺NGSパネル(8遺伝子)
ATP1A2・CACNA1S・SCN4A・KCNJ2・CLCN1・RYR1など、低カリウム性/高カリウム性周期性四肢麻痺の主要原因遺伝子8種を効率的にスクリーニング。
🤰 インペリアルプラン(NIPT・154遺伝子)
出生前にお腹のお子さんの単一遺伝子疾患リスクを評価するNIPT最広範プラン。ATP1A2を含む154遺伝子・218疾患をスクリーニング可能です。
💡 用語解説:NGSパネル検査とトリオ全エクソーム解析
NGS(次世代シーケンス)パネル検査は、特定の疾患群に関連する複数の遺伝子をまとめて解析する手法です。1遺伝子ずつ調べるよりも費用・時間・診断率すべてが効率的です。
トリオ全エクソーム解析は、患者本人と両親の3名分を同時にシーケンスする方法で、新生突然変異の同定に特に強力です。ATP1A2変異は新生変異も多いため、トリオ解析が威力を発揮します。
検査の選択は症状とご家族のニーズによって変わります。片頭痛主体ならば片頭痛パネル、てんかん主体ならば成人発症てんかんパネル、麻痺発作主体ならば周期性四肢麻痺パネルが起点になります。出生前に次のお子さんのリスク評価を希望される場合は、インペリアルプランNIPTでATP1A2を含む154遺伝子の評価が可能です。どのプランがご家族に適しているかは、臨床遺伝専門医による事前カウンセリングで一緒に検討していきます。
🔍 関連記事:遺伝カウンセリングとは遺伝子検査一覧NIPTについて
7. 治療と最新の研究動向
ATP1A2関連疾患の治療は長らく対症療法が中心でしたが、分子病態の解明とともに分子標的アプローチや既存薬の再評価、そして遺伝子治療の臨床応用が一気に進展しています。
標準的な薬物療法
- ➤カルシウムチャネルブロッカー(ベラパミル・フルナリジン):細胞内カルシウム過負荷を抑え、AHCやFHM2の発作頻度・重症度を低減します。
- ➤抗てんかん薬(バルプロ酸・ラモトリギン・レベチラセタムなど):てんかんを伴う症例に使用。アセタゾラミドも重症発作に試みられます。
- ➤急性脳症へのコルチコステロイド:脳浮腫の軽減を目的に小児で推奨されています。早期介入で重症度と持続時間を劇的に短縮できた症例報告があります。
- ➤禁忌事項:脳卒中リスクを高める血管収縮薬は避け、発作誘発リスクのある脳血管造影も推奨されません。
注目の新規アプローチ①:NMDA受容体拮抗薬メマンチン
グルタミン酸過剰による神経毒性がATP1A2脳症の中核病態であるという「グルタミン酸仮説」に基づき、グルタミン酸受容体のひとつであるNMDA受容体を阻害するメマンチンのオフラベル使用が注目されています。重度ATP1A2関連てんかん性脳症患者7名のうち5名にメマンチンが投与され、平均2.3年の追跡で全例において発作頻度の減少や歩行・協調運動・注意持続時間の改善が報告されています。
注目の新規アプローチ②:経口ATP化合物療法
画期的な治療報告として、小児交互性片麻痺(AHC)患者に対する経口ATP化合物の投与があります。フルナリジン不応のATP1A3新生変異重症AHC患者に対して、用量を21mg/kgまで増量して4年間追跡した報告では、片麻痺エピソードの頻度・持続時間の大幅な制御、歩行能力・運動スキルの明確な向上、気分状態と疲労耐性の改善が記録されました。治療を意図的に中断すると即日エピソードが再発し、再開で速やかにコントロールされたことから、外因性ATP供給がポンプ機能不全のニューロン・アストロサイトのエネルギー代謝を直接サポートする可能性が示されています。
注目の新規アプローチ③:CRISPR塩基編集による遺伝子治療
2023年末に鎌状赤血球症に対する世界初のCRISPR治療薬が承認されたことを皮切りに、希少神経疾患への応用も加速しています。ノースウェスタン大学とハーバード大学の共同研究チームは、AHC患者由来iPS細胞とAHCマウスモデルにおいて、最新の塩基編集(Base Editing)・プライム編集技術を用いてATP1A3の単一塩基変異を直接修正することに成功し、Cell誌に報告しています。送達システムとしてAAVベクターやリピドナノパーティクル(LNP)を用いた中枢神経系への遺伝子治療プラットフォームの開発も進んでおり、ATP1A2変異への応用も将来的に期待されています。
📌 治療の現在地 現時点ではメマンチンや経口ATP療法、症状別の対症療法が現実的な選択肢です。CRISPR塩基編集は前臨床段階ですが、希少神経疾患への応用は2020年代半ば以降急速に進んでおり、中長期的には根本治療の可能性が見えてきています。
8. 遺伝カウンセリングの意義
ATP1A2変異が確認された場合、ご本人・ご家族にとって重要なのは検査結果を「正しく受け止め、次の選択につなげる」プロセスです。遺伝カウンセリングでは以下のような内容を扱います。
- ➤遺伝形式と再発リスクの説明:ATP1A2のFHM2・AHC・DEE98は常染色体顕性(優性)遺伝で、ご本人がお子さんを持つ場合の遺伝確率は理論上50%。ただし重症型の多くは新生突然変異であり、ご両親は健康というケースが大半です。生殖細胞モザイクの可能性も考慮します。
- ➤表現型の幅広さの説明:同じATP1A2変異でも、家系内・家系間で軽症の片頭痛から重症脳症まで症状の幅が大きいことをお伝えします。出生前に変異が判明したからといって、必ずしも重症化するとは限りません。
- ➤出生前診断の選択肢:次のお子さんを考えていらっしゃる場合、絨毛検査・羊水検査での確定診断、またはインペリアルプランNIPTなどの選択肢があります。
- ➤長期管理と急変時の備え:発作誘因(発熱・感染・激しい運動など)の把握、緊急時の対応プラン、神経科・小児科・遺伝科の連携体制の整備をご家族と一緒に組み立てます。
💡 大切な視点 ATP1A2変異の表現型は不完全浸透・可変表現性が大きく、出生前に変異が見つかっても重症化するか軽症化するかを正確に予測することはできません。情報をどう受け止め、何を選ぶかは、最終的にご家族の価値観に委ねられる領域です。医師は中立的な立場で必要な情報をお届けすることに徹し、ご決断はご家族と共に検討します。
9. よくある誤解
誤解①「ATP1A2変異=片頭痛だけの遺伝子」
古典的な認識では片麻痺性片頭痛(FHM2)の原因でしたが、現在はAHC・DEE98・周期性四肢麻痺・心不全まで含む多臓器疾患の原因と分かっています。脳・心・筋すべての評価が必要です。
誤解②「両親が健康なら遺伝病ではない」
ATP1A2の重症型(AHC・DEE98など)は新生突然変異で発症するケースが多く、ご両親はまったく健康なことが大半です。「家族に同じ病気がいないから違う」とは判断できません。
誤解③「片頭痛だから命に関わらない」
FHM2の発作中には脳卒中と区別がつかないほど重篤になることがあり、急性重症脳症や心停止・無呼吸などの致死的イベントも報告されています。「片頭痛だから様子見」は危険です。
誤解④「変異が見つかれば確実に発症する」
ATP1A2は不完全浸透・可変表現性が大きく、同じ変異でも軽症の片頭痛のみで生涯を終える方から重症脳症まで幅があります。変異の同定は重要ですが「確実な未来予測」ではありません。
10. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 ATP1A2関連疾患の診断・遺伝カウンセリングについて
ATP1A2変異が疑われる症状について、また検査の選び方についてのご相談は、臨床遺伝専門医が在籍する東京・ミネルバクリニックにお気軽にお問い合わせください。
関連記事
参考文献
- [1] OMIM. ATP1A2 – ATPase, Na+/K+ Transporting, Alpha 2 Polypeptide. Entry #182340. [OMIM]
- [2] GeneCards. ATP1A2 Gene – ATPase Na+/K+ Transporting Subunit Alpha 2. [GeneCards]
- [3] Friedrich T, et al. ATP1A2 Mutations in Migraine: Seeing through the Facets of an Ion Pump onto the Neurobiology of Disease. Front Physiol. 2016;7:239. [PMC4914835]
- [4] Phenotypic Variability in Patients with Pathogenic Variants in ATP1A2 and ATP1A3. Neurology. 2024. [Neurology]
- [5] A novel ATP1A2 mutation in a patient with hypokalaemic periodic paralysis and CNS symptoms. J Med Genet. 2018. [PMC6262219]
- [6] Migraine-Associated Mutation in the Na,K-ATPase Leads to Disturbances in Cardiac Metabolism and Reduced Cardiac Function. J Am Heart Assoc. 2022. [PMC9075430]
- [7] Li Y, et al. Functional correlation of ATP1A2 mutations with phenotypic spectrum: from pure hemiplegic migraine to its variant forms. J Headache Pain. 2021. [PMC8359390]
- [8] Early Onset Severe ATP1A2 Epileptic Encephalopathy: Clinical Features and Response to Memantine. Pediatr Neurol. 2021. [PMC7940561]
- [9] Oral ATP treatment in alternating hemiplegia of childhood: a case report and review. Front Med. 2024. [PMC11747781]
- [10] Familial Hemiplegic Migraine. GeneReviews®. NCBI Bookshelf. [GeneReviews]
- [11] MedlinePlus Genetics. ATP1A2 gene. [MedlinePlus]
- [12] Clinical characterization of a novel ATP1A2 p.Gly615Glu mutation in nine family members with familial hemiplegic migraine. Brain Communications. 2025. [Brain Comms]






