承認済シンボル:PRPH2
遺伝子名:peripherin 2
参照:
HGNC: 9693
AllianceGenome : HGNC : 9693
NCBI:5961
Ensembl :ENSG00000112619
UCSC : uc003mke.3
遺伝子OMIM番号179605
●遺伝子のlocus type :タンパク質をコードする
●遺伝子のグループ:Tetraspanins
●遺伝子座: 6p21.1
●ゲノム座標: (GRCh38): 6:42,696,598-42,722,597
遺伝子の別名
PERIPHERIN 2, MOUSE, HOMOLOG OF
RDS, MOUSE, HOMOLOG OF
PERIPHERIN, PHOTORECEPTOR TYPE
RETINAL DEGENERATION, SLOW, MOUSE, HOMOLOG OF
RDS
peripherin-2
photoreceptor peripherin
retinal degeneration slow protein
遺伝子の概要
PRPH2(ペリフェリン2)は、光受容体の外節ディスクの形成と維持に不可欠な膜貫通タンパク質をコードする遺伝子です。この遺伝子は、網膜における視細胞の構造的完全性と機能に極めて重要な役割を果たしています。
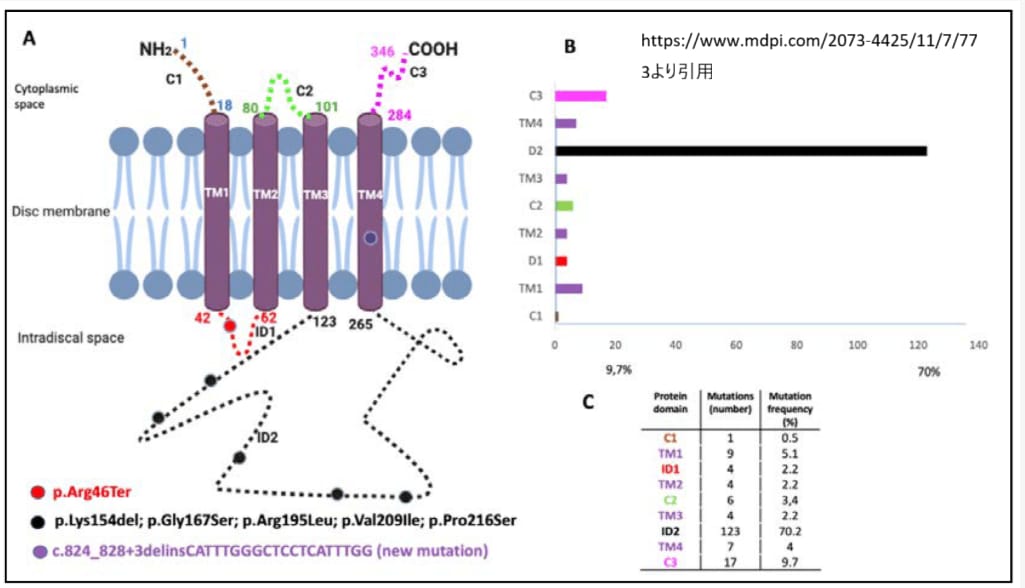
PRPH2遺伝子によってコードされるタンパク質は346アミノ酸から構成され、4つの膜貫通ドメインを持つテトラスパニン型膜タンパク質です。このタンパク質は、ROM1(網膜外節膜タンパク質1)と共に非共有結合性の四量体を形成し、さらにジスルフィド結合を介して高次のオリゴマー複合体を形成します。
光受容体細胞において、PRPH2は外節ディスクの辺縁部、すなわち円盤膜リム(disk rim)に局在し、ディスクの形態形成、安定化、圧縮に関与する接着分子として機能すると考えられています。視細胞外節は光を受容するために特化した構造で、その内部には多数の平たく潰れた袋状の円盤膜が積み重なっており、PRPH2はこれらの円盤膜の縁(外周部分)に特異的に集中しています。この円盤膜リム部は、ロドプシンなどの視物質が円盤全体に分布するのに対し、構造維持や円盤形成に関連したタンパク質が特に集まる特殊な領域となっています。
免疫蛍光法や免疫電子顕微鏡法を用いた研究により、PRPH2の細胞内局在が詳細に解析されています。野生型マウス網膜の免疫蛍光染色画像では、PRPH2(赤色)とROM1や他のマーカー(rhodopsin、opsinなど、緑色)が共染され、PRPH2が視細胞外節層に集中していることが確認されています。一方、異常変異マウスでは、PRPH2が外節層だけでなく外核層(ONL)や内節(IS)にも誤局在する様子が観察されており、高倍率画像や免疫金ラベル電子顕微鏡画像により、正常な状態ではPRPH2が外節の円盤リム全体に均一に分布していることが示されています。この機能は、視細胞における光変換過程の維持に不可欠です。
PRPH2遺伝子の変異は、網膜色素変性症、黄斑ジストロフィー、レーバー先天性黒内障など、多様な網膜変性疾患を引き起こします。興味深いことに、同じ遺伝子の異なる変異、あるいは同一の変異であっても、著しく異なる臨床表現型を示すことが知られており、遺伝子型と表現型の関係の複雑性を示しています。
さらに、PRPH2はROM1遺伝子との二遺伝子性遺伝(digenic inheritance)による網膜色素変性症の原因としても知られており、これは複数の遺伝子座の変異が組み合わさって疾患を引き起こす遺伝形式の代表的な例となっています。
遺伝子と関係のある疾患
Leber congenital amaurosis 18 レーバー先天性黒内障18 608133 AD, AR, DD 3
Macular dystrophy, patterned, 1 黄斑ジストロフィー,パターン1 169150 AD 3
Macular dystrophy, vitelliform, 3 卵黄状黄斑ジストロフィー3 608161 AD 3
Choroidal dystrophy, central areolar, 2 中心輪状脈絡膜萎縮症2 613105 AD 3
Retinitis punctata albescens 白点状網膜炎 136880 AD, AR 3
遺伝子の発現とクローニング
ヒト遺伝子
1991年、Travisらはマウスrds cDNAのコード領域由来のプローブを用いてヒト網膜cDNAライブラリーをスクリーニングし、ヒトRDS cDNAクローンを単離しました。このクローンは、マウスタンパク質と92%の相同性を持つ346アミノ酸からなる推定タンパク質をコードしていました。
ヒトとマウスのタンパク質間で、22~26残基からなる4つの疎水性ドメインと、N結合型糖鎖付加の可能性がある2つの部位が保存されていました。ノーザンブロット解析により、ヒト網膜において3.0kbと5.5kbの2つのRDS転写産物が検出されました。
Dryjaら(1989)も独立して、ヒトRDSタンパク質がマウスの類似体と92%の相同性を共有することを確認しました。
マウス遺伝子
マウスの「網膜変性、遅延型(retinal degeneration slow; rds)」は、網膜の光受容体外節の異常発生とそれに続く桿体と錐体の緩徐な変性を特徴とする表現型で、ヒトの網膜症に見られる異常に類似しています。
1989年、Travisらは、rds遺伝子が正常に光受容体で発現しているという仮説に基づいた減算cDNAクローニング戦略を用いて、この疾患の推定マウス遺伝子を単離しクローニングしました。この遺伝子は、桿体外節タンパク質-1(ROM1)と相同性を持つ346アミノ酸の網膜特異的タンパク質をコードしています。
マウスrds変異の原因は、rdsエクソン2への約10kbの外来DNA挿入によるものと考えられました。この挿入エレメント全体が変異遺伝子座のRNA産物に含まれていました。Maら(1995)は、マウスのrdsが9.2kbの反復性ゲノムエレメントの挿入変異によって引き起こされることを示し、rdsがヌル対立遺伝子であることを示す証拠を提示しました。
ウシ遺伝子
Connellら(1991)は、ウシ光受容体細胞タンパク質ペリフェリンのアミノ酸配列が、正常マウスrds遺伝子によってコードされるタンパク質の配列と92.5%同一であることを報告しました。
マッピング
Demantら(1979)は、rds変異をマウス17番染色体に局在させ、この所見はTravisら(1989)によって確認されました。
Travisら(1991)は、ヒト/ハムスター体細胞ハイブリッドパネルのDNA解析と直接in situハイブリダイゼーションにより、ヒトPRPH2遺伝子が6番染色体短腕の近位部に位置することを示しました。
現在の精密なゲノム座標(GRCh38)では、PRPH2遺伝子は6p21.1、具体的には6:42,696,598-42,722,597に位置することが確認されています。
遺伝子の機能
PRPH2とROM1は、非共有結合性四量体およびジスルフィド結合による高次オリゴマーを形成するテトラスパニン膜タンパク質であり、光受容体ディスク形態形成に関与しています。
タンパク質の構造と局在
Bascomら(1990)は、RDS遺伝子産物を分子および超微細構造レベルで特徴づけるための実験を行いました。予備的証拠により、RDSとROM1遺伝子産物がin vivoでジスルフィド結合を用いてヘテロ二量体を形成することが示されました。
Travisら(1991)は、予測タンパク質配列から誘導された合成ペプチドに対する抗体を用いて、rdsタンパク質が光受容体外節ディスクに限定された膜結合型糖タンパク質であることを示しました。このタンパク質は、外節ディスクの安定化と圧縮に関与する接着分子として機能する可能性があります。
Connellら(1991)は、モノクローナル抗体とウエスタンブロット解析を用いて、rds変異体で欠損しているペリフェリンタンパク質が、正常では桿体外節に局在し、1つ以上のジスルフィド結合によって連結された2つのサブユニットとして存在することを示しました。
オリゴマー形成と機能
Loewen ら(2001)は、L185P変異体(二遺伝子性網膜色素変性症に関連)が自己組織化して二量体を形成し、さらに分子間ジスルフィド結合を介して四量体を形成するが、野生型ペリフェリン-2に特徴的な高次オリゴマーは形成しないことを示しました。しかし、L185P変異体は野生型ROM1および野生型ペリフェリン-2と相互作用してコア四量体および高次ジスルフィド結合オリゴマーを形成することができました。
輸送と極性化
Leeら(2006)は、複数のノックアウトおよびトランスジェニック動物モデルにおいて、ペリフェリン/rdsとRom1の輸送を研究しました。ペリフェリン/rdsの輸送と局在は、発生中の光受容体においてディスク形成前に外節形態形成部位に極性化されていました。
ペリフェリン/rdsとRom1の輸送は、ロドプシンノックアウトマウスでも維持されており、リム(rim)タンパク質とロドプシンが別々の輸送経路を持つことが示唆されました。短縮型ペリフェリン/rds-GFPの外節での存在は、ペリフェリン/rdsマウスがホモ四量体を形成して外節をターゲットにするという以前の証拠を支持しました。
Rom1がrdsマウスの外節ドメインに輸送されるという発見は、Rom1が独自の選別および輸送シグナルを有する可能性を示唆しました。
分子遺伝学
網膜色素変性症7(RP7)
Farrarら(1991)は、常染色体優性網膜色素変性症-7(RP7)の大規模なアイルランド家系において、RDS遺伝子の3塩基対欠失を同定しました。この欠失により、ペリフェリンの予測される第3膜貫通ドメインにある高度に保存されたシステイン残基対(コドン118または119)の1つが失われました。この欠失は疾患表現型と共分離しましたが、非罹患者には存在しませんでした。
Kajiwaraら(1991)は、網膜色素変性症患者4人において、RDS遺伝子のヘテロ接合性変異を同定しました。変異には、プロリン219を正確に削除する3塩基対欠失、プロリン216をロイシンに変える点変異、およびロイシン185をプロリンに置換する変異が含まれていました。
二遺伝子性網膜色素変性症
Kajiwaraら(1994)は、RDS遺伝子のL185P変異がROM1遺伝子のヌル変異と組み合わさった二重ヘテロ接合状態でのみ網膜色素変性症を引き起こすことを実証しました。これは、疾患のメカニズムとして2遺伝子座仮説の分子的証明の初期の例の1つでした。
Kedzierskiら(2001)は、一連のトランスジェニック/ノックアウトマウス実験を行い、マウスモデルにおける二遺伝子性RPの臨床観察を裏付けました。二遺伝子性RPのマウスモデルにおける光受容体変性は、野生型および単一遺伝子コントロールよりも速く進行しました。RDSとROM1タンパク質の単純な欠乏が、RDS媒介RPにおける光受容体変性の原因と考えられました。
黄斑ジストロフィー
Wellsら(1993)は、様々な黄斑ジストロフィーを有する13人のプロバンドのPRPH2遺伝子を解析し、中心網膜に影響する黄斑ジストロフィー(中心輪状脈絡膜萎縮症-2; CACD2)を有する3家系の罹患メンバーにおいてコドン172のヘテロ接合性ミスセンス変異(R172Q, R172W)を同定しました。また、成人発症卵黄状黄斑ジストロフィー(VMD3)の女性においてヘテロ接合性ナンセンス変異も同定されました。
Travis and Hepler(1993)は、RDS遺伝子の変異によって引き起こされる表現型的に異なる網膜疾患の多様性についてコメントしました。変異は桿体と錐体の両方に影響を及ぼすようです。一部の変異は常染色体優性網膜色素変性症を伴いますが、他の変異は黄斑ジストロフィー、白点状網膜炎、または中心窩のパターン色素ジストロフィーの表現型を示します。
中心輪状脈絡膜萎縮症(CACD2)
Hoyngら(1996)は、CACD2を有する7家系のPRPH2遺伝子エクソン1を解析し、各家系の罹患者においてarg142-to-trp(R142W)変異のヘテロ接合性を同定しました。この変異は、20/20視力を有し、眼底検査や蛍光眼底造影で後極部異常のない65歳の女性家族メンバーでも検出されました。
Boonら(2009)は、オランダのCACD患者103人を解析し、45の異なる家系からの98人の患者でR142W変異を、1家系の5人の罹患家族メンバーでR172Q変異を同定しました。R142W保有CACD患者の大多数はオランダ南東部地域出身で、ハプロタイプ解析は共通の創始者変異を示唆しました。
レーバー先天性黒内障18(LCA18)
Wangら(2013)は、既知のLCAまたは若年性RP遺伝子の変異が陰性であった早期発症網膜ジストロフィーの3人の無関係な患者において、PRPH2遺伝子のホモ接合性変異を同定しました。2人の患者(1人はLCA18と診断、もう1人は若年性RPと診断)は、以前に二遺伝子性RP7患者で検出されたL185P変異のホモ接合体でした。3人目の患者(LCAと診断)は、PRPH2の別のミスセンス変異(C213R)のホモ接合体でした。
表現型の多様性
Manesら(2015)は、主にフランス出身の常染色体優性RPと診断された310家系のコホートをスクリーニングし、32のプロバンドで15の異なるPRPH2変異を同定しました。これらの家系からの27~67人の患者の臨床所見を研究し、多様な表現型を同定しました。
一部の患者は、正常、中等度低下、または重度低下した視力を伴う黄斑病変を有していました。一部は、網膜周辺部に少数の萎縮斑を伴い黄斑が温存された軽度RPを示しました。他の症例では、中周辺網膜に典型的な色素沈着とびまん性萎縮が認められました。一部の患者は病変の傍中心局在を示し、他の家族メンバーはびまん性のRP形態を有していました。1家系では、母親が典型的なRPを有し、息子はRPの徴候なしに卵黄状中心窩沈着物を有していました。
遺伝子型と表現型の相関
Keen and Inglehearn(1996)は、ヒトRDS遺伝子において合計43の配列変異が記載されていることを報告しました。これには、30のミスセンス変異、終止コドンを生成する2つの1塩基置換、7つの小さなインフレーム欠失、および読み枠を破壊する4つの挿入/欠失イベントが含まれていました。
これらのうち39は、4つの広いカテゴリーに分類できる網膜表現型と関連していました:優性網膜色素変性症、進行性黄斑変性、二遺伝子性RP、およびパターンジストロフィー。
優性RPおよび重症黄斑変性の基礎となる変異は、主に第3と第4膜貫通ドメイン間の大きなディスク内ループにおけるミスセンスまたは小さなインフレーム欠失でした。対照的に、軽度のパターン表現型または二遺伝子性RPと関連する変異は、遺伝子全体により均等に散在しており、しばしばナンセンス変異でした。
Keen and Inglehearn(1996)は、この区別が、大きなループがRDS分子間およびディスク内の他のタンパク質成分との相互作用の重要な部位であるという仮説を支持すると述べました。
Kohlら(1997)は、主に中心性網膜ジストロフィーの様々な形態を有する76の独立した家系において、RDS遺伝子の変異をスクリーニングしました。2つのナンセンス変異、5つのミスセンス変異、および1つの1塩基挿入が検出されました。これらすべてがヘテロ接合状態でした。Kohlらは、家系間および家系内での表現型と疾患発現の顕著な変動についてコメントしました。
Anandら(2009)は、PRPH2遺伝子のR172Q、R172W、およびR142W変異を有する患者における地図状萎縮の総面積と年齢関連視力データを解析しました。R172W変異は、R142WまたはR172Qと比較して、より早期の発症年齢とより悪い視力への傾向が観察されました。
線形回帰分析により、60歳までの視力はR172W変異ではR142W(p<0.001)またはR172Q(p=0.04)変異よりも有意に悪いことが示されました。これらの所見は、PRPH2遺伝子の変異に関連する視覚予後が変異特異的である可能性があることを示唆しています。
動物モデル
マウスモデル
Maら(1995)は、マウスのrds表現型が、rdsエクソン2への9.2kbの反復性ゲノムエレメントの挿入変異によって引き起こされることを示しました。この挿入エレメントはH-2複合体のハプロタイプ特異的エレメントと非常に類似しています。全エレメントが変異遺伝子座のRNA産物に含まれていました。Maらは、マウスのrdsがヌル対立遺伝子を表すという証拠を提示しました。
遺伝子治療研究
Aliら(2000)は、Prph2トランスジェンをコードする組換えアデノ随伴ウイルスの網膜下注射により、外節構造の安定的生成と、ペリフェリン-2とロドプシンの両方を含む新しいディスクスタックの形成がもたらされることを実証しました。多くの場合、これらは形態学的に正常な外節と類似していました。
さらに、光受容体層の構造的完全性の再確立により、電気生理学的矯正がもたらされました。これらの研究は、in vivo遺伝子導入による複雑な超微細構造の矯正が初めて実証されたものでした。
Sarraら(2001)は、超微細構造改善の可能性は動物が治療される年齢に依存するが、単回注射の光受容体超微細構造への効果は長期的である可能性があることを実証しました。しかし、投与日にかかわらず、形態と機能の改善にもかかわらず、光受容体細胞喪失に対する有意な効果はありませんでした。
これらの所見は、光受容体欠損を有する患者における成功的な遺伝子治療は、最終的には疾患の早期段階での介入とトランスジェン発現の正確な制御に依存する可能性があることを示唆しました。
変異特異的モデル
McNallyら(2002)は、マウスにおいてrds-ペリフェリン遺伝子のコドン307に標的単塩基欠失を導入しました。これは、Apfelstedt-Syllaら(1995)によって報告されたヒト変異に類似しています。組織病理学的および網膜電図解析により、コドン307変異のヘテロ接合体およびホモ接合体マウスにおける網膜症は、rds +/-およびrds -/-マウスよりも重症であるように見え、rds-307変異が表現型に対して優性負性効果を発揮する可能性があることが示唆されました。
Dingら(2004)は、RdsにR172W変異を保有するトランスジェニックマウスを作製しました。変異Rdsは適切に局在していましたが、トランスジェン発現レベルと表現型の発症/重症度の間に直接的な相関が存在しました。野生型背景では、錐体および桿体光受容体の両方の構造と機能が有意に減弱し、優性負性錐体-桿体欠損が示唆されました。
R172W/Rds二重ヘテロ接合体マウスでは、錐体応答は野生型レベルの40%に減弱し、変異の錐体への優先的損傷効果が示されました。逆に、R172W/Rds二重ヘテロ接合体は、桿体機能の有意な救済と桿体外節構造の改善を示しました。
Chakrabortyら(2009)は、Rds C150S変異を含むトランスジェニックマウスが、高次Rdsオリゴマーの形成に失敗することを示しました。C150S-Rdsマウスは、著明な早期発症の錐体機能低下と異常な外節構造を示しました。対照的に、桿体におけるC150S-Rds発現は、Rds +/-表現型を部分的に救済しました。
桿体と錐体の見かけの外節構造の違いは、錐体が高次Rds/Rom1オリゴマーの排除(例えば、Rds C150残基の変異によって媒介される)に対してより感受性が高い可能性があることを示唆しています。
錐体特異的研究
野生型マウス光受容体の約3%のみが錐体であるため、R172W変異を保有するトランスジェニックマウスの錐体をよりよく研究するために、Conleyら(2014)は、発生中の桿体が錐体様運命を採用するNrl -/-マウス背景を使用しました。R172W変異が錐体外節の超微細構造に異常を引き起こすこと、および異常に大きなRom1複合体の形成を引き起こすことが明らかになりました。
スケートモデル
Liら(2003)は、哺乳類ペリフェリン/rdsのスケートオルソログを同定しました。ヒト網膜疾患に関連する残基の大部分の保存は、これらの残基が重要な機能的役割を果たすことを示しています。
アレリックバリアント
アレリック・バリアント(24例選択):Clinvarはこちら
- .0001 網膜色素変性症7
- PRPH2, CYS118DEL
常染色体優性網膜色素変性症-7の大規模なアイルランド家系において、Farrarら(1991)はRDS遺伝子の3塩基対欠失を同定し、ペリフェリンの予測される第3膜貫通ドメインにある高度に保存されたシステイン残基対(コドン118または119)の1つが失われる結果となりました。 - .0002 網膜色素変性症7
- PRPH2, 3-BP DEL, PRO219DEL
常染色体優性網膜色素変性症-7の患者において、Kajiwaraら(1991)はRDS遺伝子の3塩基対欠失を同定し、正常ではプロリンを指定するコドン219が正確に削除されました。 - .0003 網膜色素変性症7
- PRPH2, PRO216LEU
常染色体優性網膜色素変性症-7の単一患者において、Kajiwaraら(1991)はRDS遺伝子のC-T転移を同定し、プロリン216からロイシンへの変化(P216L)をもたらしました。 - .0004 網膜色素変性症7、二遺伝子性/レーバー先天性黒内障18/パターン黄斑ジストロフィー1
- PRPH2, LEU185PRO
ロドプシン変異が陰性であった139人のRP患者のコホートから常染色体優性網膜色素変性症の無関係な患者2人において、Kajiwaraら(1991)はRDS遺伝子のヘテロ接合性T-C転移を同定し、ロイシン185からプロリンへの置換(L185P)をもたらしました。Kajiwaraら(1994)は、L185P変異がROM1遺伝子のヌル変異と組み合わさった二重ヘテロ接合状態でのみ網膜色素変性症を引き起こすことを実証しました。Wangら(2013)は、早期発症網膜ジストロフィーの2人の患者において、PRPH2遺伝子のL185P変異のホモ接合性を同定しました。 - .0005 白点状網膜炎、常染色体優性
- PRPH2, 2-BP DEL
黄斑を含む進行性網膜変性および周辺網膜下白斑(白点状網膜炎)を伴う59歳男性において、Kajiwaraら(1992、1993)はRDS遺伝子のコドン25の2塩基対欠失を同定し、その下流54塩基で早期終止コドンに至りました。 - .0006 中心輪状脈絡膜萎縮症2
- PRPH2, ARG172GLN
中心網膜に影響する常染色体優性黄斑ジストロフィーを分離する家系の罹患メンバー4人において、Wellsら(1993)はPRPH2遺伝子のG-A転移を同定し、アルギニン172からグルタミンへの変化(R172Q)をもたらしました。 - .0007 中心輪状脈絡膜萎縮症2
- PRPH2, ARG172TRP
中心網膜に影響する常染色体優性黄斑ジストロフィーを分離する2家系の罹患メンバーにおいて、Wellsら(1993)はPRPH2遺伝子のヘテロ接合性C-T転移を同定し、アルギニン172からトリプトファンへの置換(R172W)をもたらしました。Payneら(1998)は、常染色体優性黄斑ジストロフィーを分離する11のイギリス家系でR172W変異を同定しました。 - .0008 卵黄状黄斑ジストロフィー3
- PRPH2, TYR258TER
成人卵黄状黄斑ジストロフィーの女性において、Wellsら(1993)はチロシン258を含むナンセンス変異(Y258X)を同定しました。 - .0009 パターン黄斑ジストロフィー1
- PRPH2, GLY167ASP
Nicholsら(1993)は、3世代12人が網膜色素上皮のバタフライジストロフィーを有する家系を研究しました。罹患者において、RDS遺伝子のG-A転移が見つかり、グリシン167からアスパラギン酸への置換(G167D)をもたらしました。 - .0010 パターン黄斑ジストロフィー1
- PRPH2, 2-BP DEL, 1137TG
Nicholsら(1993)は、中心窩のバタフライ型色素ジストロフィーを有する家系の罹患メンバーにおいて、RDS遺伝子のコドン299と300に重なる2塩基対の欠失を記述しました。 - .0011 網膜色素変性症7
- PRPH2, ASN244LYS
Kikawaら(1994)は、常染色体優性網膜色素変性症の一形態を有する家系において、RDS遺伝子のアスパラギン244からリジンへの変異(N244K)を同定しました。 - .0012 卵黄状黄斑ジストロフィー3
- PRPH2, PRO210ARG
成人発症中心窩黄斑ジストロフィーおよび脈絡膜新生血管を有する男性において、Feistら(1994)はPRPH2遺伝子のプロリン210からアルギニンへの変異(P210R)のヘテロ接合性を同定しました。 - .0013 パターン黄斑ジストロフィー1
- PRPH2, 4-BP INS, CODON 140
Kimら(1995)は、おそらく常染色体優性遺伝を示す網膜のパターンジストロフィーを有する血統を研究し、ペリフェリン/RDS遺伝子のコドン140に4塩基対挿入を実証しました。 - .0014 卵黄状黄斑ジストロフィー3
- PRPH2, MET1THR
成人発症卵黄状黄斑ジストロフィーの67歳ドイツ人患者において、Felborら(1997)はPRPH2遺伝子のメチオニン1からスレオニンへのアミノ酸置換(M1T)を同定しました。 - .0015 卵黄状黄斑ジストロフィー3
- PRPH2, TRP316TER
成人卵黄状黄斑ジストロフィーの55歳ドイツ人患者において、Felborら(1997)はRDS遺伝子のトリプトファン316から終止コドンへのナンセンス変異(W316X)を同定しました。 - .0016 卵黄状黄斑ジストロフィー3
- PRPH2, 1-BP DEL, 112G
Yangら(2003)は、脈絡膜新生血管を伴う成人発症中心窩黄斑ジストロフィーに罹患した8人の家族メンバー全員が、RDS遺伝子のフレームシフトヌル変異、ヌクレオチド112の1塩基対欠失(112delG)を有することを報告しました。 - .0017 網膜色素変性症7/パターン黄斑ジストロフィー1
- PRPH2, 3-BP DEL
Weleberら(1993)は、RDS遺伝子のコドン153または154の3塩基対欠失を有する単一家系内での3つの別々の表現型の発生を報告しました。 - .0018 網膜色素変性症7
- PRPH2, ARG46TER
網膜色素変性症の58歳ドイツ人男性において、Meinsら(1993)はRDS遺伝子エクソン1のヘテロ接合性アルギニン46から終止コドンへの変異(R46X)を同定しました。 - .0019 網膜色素変性症7
- PRPH2, 1-BP DEL, 1160T
典型的な遅発性網膜色素変性症を有する68歳ドイツ人女性において、Gruningら(1994)はRDS遺伝子のコドン307の1塩基対欠失を同定しました。 - .0020 網膜色素変性症7
- PRPH2, ASP173VAL
大規模多世代スペイン家系の網膜色素変性症-7の患者10人において、Gruningら(1994)はRDS遺伝子のA-T転換を同定し、アスパラギン酸173からバリンへの置換(D173V)をもたらしました。 - .0021 中心輪状脈絡膜萎縮症2
- PRPH2, ARG195LEU
黄斑部の明確に境界された進行性脈絡網膜萎縮を伴う日本人家系の罹患メンバー3人において、Yanagihashiら(2003)はPRPH2遺伝子の転換のヘテロ接合性を同定し、アルギニン195からロイシンへの置換(R195L)をもたらしました。 - .0022 中心輪状脈絡膜萎縮症2
- PRPH2, ARG142TRP
中心輪状脈絡膜萎縮症を有する7家系からの11人の罹患メンバーにおいて、Hoyngら(1996)はPRPH2遺伝子エクソン1の664C-T転移のヘテロ接合性を同定し、アルギニン142からトリプトファンへの置換(R142W)をもたらしました。 - .0023 パターン黄斑ジストロフィー1/レーバー先天性黒内障18
- PRPH2, CYS213ARG
パターン黄斑ジストロフィーの患者において、Payneら(1998)はPRPH2遺伝子のシステイン213からアルギニンへの置換(C213R)のヘテロ接合性を同定しました。Wangら(2013)は、レーバー先天性黒内障と診断された早期発症網膜ジストロフィーの29歳女性において、C213R置換をもたらすPRPH2遺伝子のホモ接合性を同定しました。 - .0024 卵黄状黄斑ジストロフィー3/パターン黄斑ジストロフィー1
- RDS, TYR141CYS
成人発症中心窩黄斑ジストロフィーを有する2家系およびパターン黄斑ジストロフィーを有する1家系からの罹患者において、Yangら(2004)はRDS遺伝子のヘテロ接合性c.422A-G転移を同定し、ディスク内空間におけるチロシン141からシステインへの置換(Y141C)をもたらしました。
よくあるご質問
- PRPH2遺伝子の変異はどのような疾患を引き起こしますか?
- PRPH2遺伝子の変異は、網膜色素変性症、黄斑ジストロフィー、レーバー先天性黒内障、中心輪状脈絡膜萎縮症、白点状網膜炎など、多様な網膜変性疾患を引き起こします。同じ遺伝子の異なる変異、あるいは同一の変異であっても、家系や個人により表現型が大きく異なることが特徴です。
- 二遺伝子性遺伝(digenic inheritance)とは何ですか?
- 二遺伝子性遺伝とは、2つの異なる遺伝子座の変異が組み合わさって初めて疾患を発症する遺伝形式です。PRPH2遺伝子とROM1遺伝子の変異を両方持つ場合に網膜色素変性症を発症する例が、この遺伝形式の代表的な事例として知られています。それぞれ単独の変異では発症しない場合があります。
- PRPH2遺伝子変異による疾患の遺伝形式は?
- PRPH2遺伝子変異による疾患は、主に常染色体優性遺伝の形式をとりますが、常染色体劣性遺伝や二遺伝子性遺伝の形式をとる場合もあります。ヘテロ接合性変異(片方の遺伝子のみの変異)で発症する場合が多いですが、ホモ接合性変異(両方の遺伝子の変異)ではより重症の早期発症型となることがあります。
- 同じ家系内でも症状が異なることはありますか?
- はい、PRPH2遺伝子変異の特徴として、同じ家系内、さらには同一の変異を持つ家族間でも臨床症状が大きく異なることがあります。例えば、ある家系では網膜色素変性症、パターン黄斑ジストロフィー、黄斑変性など、異なる表現型を示すことが報告されています。この表現型の多様性には、遺伝的修飾因子や環境要因が関与していると考えられています。
- PRPH2遺伝子変異の検査はどのような場合に推奨されますか?
- 網膜色素変性症、黄斑ジストロフィー、若年発症の視力障害、家族歴のある網膜疾患などが疑われる場合に検査が推奨されます。特に、視野狭窄、夜盲、中心視力の低下、特徴的な眼底所見(色素沈着、黄斑部の異常など)がある場合は、PRPH2遺伝子を含む網膜疾患関連遺伝子の検査を検討します。
- PRPH2遺伝子変異に対する治療法はありますか?
- 現時点では根本的な治療法は確立されていませんが、研究段階では遺伝子治療の可能性が示されています。動物モデルでは、正常なPRPH2遺伝子を導入することで光受容体の構造と機能が部分的に回復することが報告されています。臨床的には、ロービジョンケア、遮光眼鏡の使用、補助具の活用などの対症療法が中心となります。定期的な眼科検査により疾患の進行をモニタリングすることが重要です。
- 家族に遺伝子変異が見つかった場合、どうすればよいですか?
- 家族内でPRPH2遺伝子変異が見つかった場合は、遺伝カウンセリングを受けることをお勧めします。血縁者も同じ変異を持つ可能性があるため、症状の有無にかかわらず眼科検査と遺伝子検査を検討することが推奨されます。変異保有者であっても症状が軽度または無症状の場合もあるため、定期的なフォローアップが重要です。







