承認済シンボル:DHCR7
遺伝子名:7-dehydrocholesterol reductase
参照:
HGNC: 2860
AllianceGenome : HGNC : 2860
NCBI:1717
Ensembl :ENSG00000172893
UCSC : DHCR7 (ENST00000355527.8) from GENCODE V46
遺伝子OMIM番号602858
●遺伝子のlocus type :タンパク質をコードする
●遺伝子のグループ:
●遺伝子座:
●ゲノム座標:
遺伝子の別名
D7SR
delta-7-dehydrocholesterol reductase
DHCR7_HUMAN
sterol delta-7-reductase
遺伝子の概要
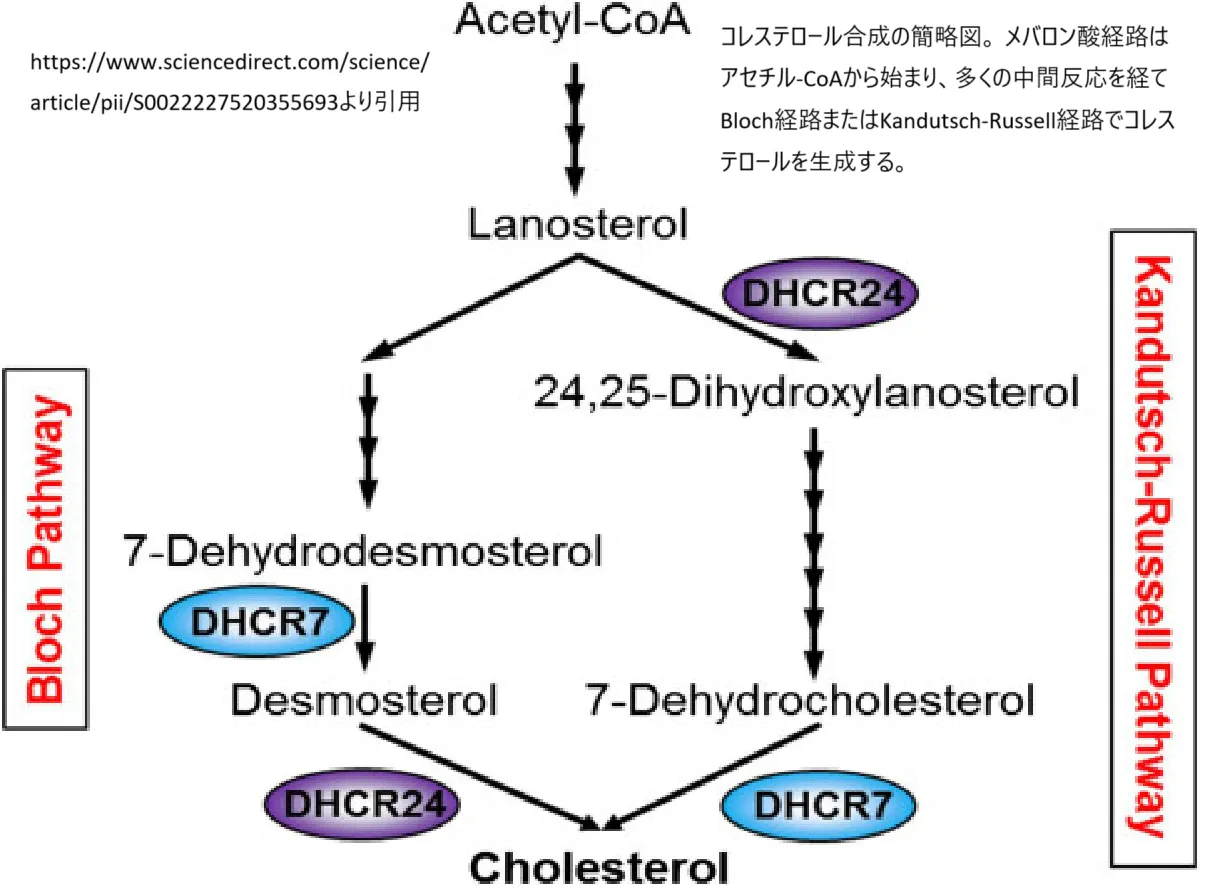
遺伝子の発現とクローニング
Waterhamら(1998年)は、シロイヌナズナの酵素のアミノ酸配列を基にESTデータベースを検索し、ヒトの7-デヒドロコレステロール還元酵素をコードする部分転写物を特定しました。さらに、5′-RACE法を用いて完全な配列を単離し、Saccharomyces cerevisiae(酵母)に異種発現させることで、この遺伝子が7-デヒドロコレステロール還元酵素をコードしていることを確認しました。
Moebiusら(1998年)は、推定される分子量55kDのDHCR7タンパク質をクローニングし、このタンパク質が6~9個の膜貫通領域を持つ膜結合型であることを示しました。植物や酵母のステロール還元酵素と類似しており、ヒトでは全身で発現し、特に副腎、肝臓、精巣、脳で多く見られます。酵母における異種発現では、7-デヒドロコレステロールからコレステロールへの変換がNADPH依存で進行し、この酵素は特定の化合物によって強力に阻害されることが確認されました。
マッピング
放射線による染色体の断片化:まず、ヒトのような目的の細胞に放射線を照射し、染色体をランダムに断片化させます。
細胞融合:次に、放射線で断片化されたヒト細胞を、染色体が欠損しているハムスターなどの異種の細胞と融合させ、ハイブリッド細胞を作成します。このハイブリッド細胞には、ヒトの染色体断片がランダムに取り込まれます。
マッピング:複数のハイブリッド細胞株を作成し、それらの細胞で特定の遺伝子やDNA断片の存在を調べます。各細胞株に含まれている染色体断片の情報を基に、対象遺伝子がどの染色体のどの位置にあるかを推定します。
放射線ハイブリッドマッピング法は、遺伝子を比較的高精度に染色体上に位置づけることができるため、遺伝子マッピングやゲノム解析に広く利用されています。この手法によって、物理的な遺伝子の位置や距離を解析することが可能です。
一方、Waterhamら(1998年)は、FISH法を用いて、DHCR7遺伝子を11q13に割り当てました。
また、Fitzkyら(1998年)は、ヒトおよびマウスのDHCR7遺伝子を詳細に解析し、FISH法により、ヒトでは11q13に、マウスでは相同領域の7F5にマッピングしました。これらの研究により、DHCR7遺伝子の染色体上の位置が明確にされ、コレステロール代謝に関与する重要な遺伝子としての役割が明らかになりました。
遺伝子の機能
さらに、Freitasら(2024年)およびLiら(2024年)は、DHCR7をプロフェロトーシス遺伝子として特定しました。フェロトーシスは、鉄依存性の細胞死の一種です。DHCR7のノックアウトは、7-デヒドロコレステロール(7-DHC)の蓄積を引き起こし、細胞をフェロトーシスに対して耐性のある状態にしました。7-DHCの蓄積は、リン脂質の過酸化を抑制し、細胞を保護する働きを持つことが確認されました。この作用は、腫瘍の増殖や腎臓の虚血再灌流障害からの保護にも関与していると示唆されています。DHCR7の再発現により、7-DHCは減少し、細胞は再びフェロトーシスに感受性を示すようになりました。
分子遺伝学
SLOSの患者は、重度の神経発達遅延、小頭症、外観異常(口唇裂、眼瞼裂斜下、小顎症など)、内臓の奇形(心臓や肺などの異常)を伴うことが多いです。Sheferら(1995年)によると、SLOS患者の肝臓ミクロソームでは、DHCR7の酵素活性が低下していることが示されており、コレステロール不足と7-DHCの蓄積がこれらの症状に関与しています。
Wassifら(1998年)、Fitzkyら(1998年)、Waterhamら(1998年)などの研究により、SLOS患者におけるDHCR7遺伝子の変異が多数特定されており、これらの変異の中にはスプライス部位変異やミスセンス変異が含まれます。これらの変異が酵素の機能を損ない、病態を引き起こします。
Witsch-Baumgartnerら(2000年)やYuら(2000年)の研究では、SLOSの遺伝的多様性に関する情報が収集され、40種類以上の異なるDHCR7遺伝子変異が特定されました。特に、IVS8-1G-C変異が多くのSLOS患者で一般的に見られることが報告されています。この変異は、エクソン9の異常なスプライシングや、フレームシフトを引き起こし、タンパク質の機能喪失をもたらします。
また、Krakowiakら(2000年)やWitsch-Baumgartnerら(2001年)は、SLOSの重症度は、コレステロールレベルや遺伝子変異の種類と関連していることを示し、変異が疾患の進行や症状の出現にどのように影響するかを研究しました。
動物モデル
さらに、Kovarovaら(2006年)は、Dhcr7 -/- マウスの肥満細胞における異常を発見しました。これらの細胞は、Fcer1受容体の刺激後に過剰な脱顆粒とサイトカインの恒常的産生を示しました。7-DHCが脂質ラフトに蓄積することで、ラフトの安定性が部分的に損なわれ、Lynキナーゼの移動や活性が異常を呈し、Lyn依存性シグナル伝達の低下とAktのリン酸化増加が観察されました。この研究は、SLOS患者におけるアレルギー感受性の増加が、脂質ラフトの機能不全によるものである可能性を示唆しています。
アレリックバリアント
DHCR7、IVS8AS、G-C、-1
DHCR7遺伝子の第8イントロンにおけるG-Cトランスバージョンは、スプライス部位の欠陥とヌル変異を引き起こし、スミス・レムリ・オピッツ症候群(SLOS; 270400)(Wright et al., 2003)で最も一般的な変異アレルです。
重症SLOS患者由来の細胞株A2SLOにおいて、Wassif ら(1998年)はDHCR7遺伝子のヌクレオチド788と789の間に134bpの挿入を同定しました。この挿入により、アミノ酸263から開始するフレームシフトが生じ、高度に保存されたカルボキシル末端のタンパク質の翻訳が妨げられました。この細胞株は挿入のホモ接合型でした。
Waterham ら(1998年)は、血縁関係のない両親の第一子である重度のSLOS患者において、DHCR7遺伝子内のヌクレオチド963番目以降に134bpの挿入変異が認められるホモ接合型を発見しました。Yu ら(2000年)は、IVS8-1G-C 変異により、最後のエクソンであるエクソン9の異常なスプライシングが起こり、イントロン8の配列が134bp挿入され、結果としてフレームシフトが起こり、早期に翻訳が停止することを指摘しています。この患者のより重篤な表現型は、挿入がコレステロール還元酵素間で強く保存されている領域で起こったという事実と一致しています。両親はともに挿入のヘテロ接合型でした。Waterham ら (1998) は、SLOS の子供において、134-bp の挿入が複合ヘテロ接合状態で発見され、もう一方のアレルには DHCR7 遺伝子における trp248-to-cys 変異 (602858.0008) が認められました。
Witsch-Baumgartner ら(2000年)は、重症のSLOS患者16人(重症度スコア50以上)を対象に、32の変異アレルのうち14のアレルにDHCR7遺伝子におけるIVS8-1G-C変異が認められることを発見しました。
Yu ら(2000年)は、この突然変異を迅速に検出するための、PCR に基づく制限エンドヌクレアーゼ消化アッセイを報告しており、66のアレリックのうち21で同定されています。 著者らは、IVS8-1G-C 転換は米国のSLOS患者において非常に一般的な突然変異であると結論づけています。
Witsch-Baumgartner ら(2001年)は、英国、ドイツ、ポーランド、オーストリアの59人のSLOS患者を対象とした研究で、118のアレリックのうち25(21.2%)でIVS8-1G-C変異を検出しました。 東西の勾配が検出されました(英国ではアレリックの34.1%、ドイツ/オーストリアでは20.5%、ポーランドでは3.3%)。DHCR7遺伝子のコーディング配列における8つの一塩基多型を用いたハプロタイプ分析により、13のIVS8-1G-C染色体が同じハプロタイプを共有していることが示され、創始者効果の証拠が示されました。1つのIVS8-1G-C変異は、異なるハプロタイプ上のT93M変異(602858.0009)とcisで存在していました。もう一方のT93M対立遺伝子もこのハプロタイプによって特徴づけられていたため、二重変異は組み換え事象と一致していました。
Krakowiakら(2000年)は、SLOS患者である2人の兄弟において、この変異をthr289からile(602858.0015)との複合ヘテロ接合性で検出しました。Nowaczyk ら(2001年)は、この兄弟の女性従姉妹もIVS8-1G-C/T289I遺伝子型を有していることを発見しました。 2人の無関係な母親は、IVS8-1G-C変異のキャリアでした。
Nowaczyk ら(2001年)は、重症のSLOSおよび全前脳症の胎児と新生児2人のホモ接合型でIVS8-1G-C変異を発見しました。 彼らは、この変異のホモ接合型である重症のSLOS患者として以前に報告された6人の新生児のうち、全前脳症を患っていた新生児はいないと述べています。
IVS8-1G-C 変異は最も頻繁に確認される変異アレリックであり、報告されたSLOSアレリックの約3分の1を占めています。Wright ら(2003)は、アフリカ系アメリカ人におけるこの変異のスクリーニングを行い、キャリア頻度は0.73%(1378人中10人)であることを発見しました。彼らは、IVS8-1G-C対立遺伝子が混血を通じてアフリカ系アメリカ人集団に現れた可能性が高いと示唆しました。Witsch-Baumgartnerら(2001年)は、この突然変異はブリテン諸島で生じた創始者変異である可能性を示唆しました。
Scalco ら(2005年)は、ブラジル人SLOS患者14人を対象とした研究で、28のアレルのうち10個(36%)にIVS8-1G-C変異を検出しました。7人の患者では、IVS8-1G-C変異がT93M変異との複合ヘテロ接合性で発見されました(602848.0009)。
Witsch-Baumgartner ら(2008年)は、ヨーロッパのアレルのハプロタイプ解析とチンパンジーの Dhcr7 相同遺伝子との比較により、彼らが 964-1G-C と呼ぶ IVS8-1G-C 変異は約 3,000 年前に北西ヨーロッパで出現したと推定しました。
0.0002 スミス・レムリ・オピッツ症候群
DHCR7、96-BP欠失
ワッシフらは(1998年)、重度のスミス・レムリ・オピッツ症候群(SLOS;270400)患者由来の細胞株(B3SLO)が、96bpの欠失とヌクレオチド505と506の間のシトシンの挿入による複合ヘテロ接合であることを発見しました(602858.0003)。欠失はヌクレオチド -77 からヌクレオチド 19 まで広がっていました。これにより開始コドンが削除される可能性がありました。欠失していない5’領域に存在する2つのATGはどちらもインフレームではなく、次のインフレームATGはアミノ酸138をコードしていました。1bpの挿入により、アミノ酸170から始まるフレームシフトと、39個の異常アミノ酸の付加が起こりました。
0.0003 スミス・レムリ・オピッツ症候群
DHCR7、1-bp 挿入、505C
重症スミス・レムリ・オピッツ症候群(SLOS)患者(270400)において複合ヘテロ接合状態で発見されたDHCR7遺伝子における1bp挿入(505_506insC)に関するWassifらの研究(1998年)については、602858.0002を参照してください。
.0004 スミス・レムリ・オピッツ症候群
DHCR7, 1-BP INS, 586T
重症スミス・レムリ・オピッツ症候群(SLOS; 270400)患者由来の細胞株C4SLOにおいて、Wassif ら(1998)は、ヌクレオチド586と587の間にチミジンが1つ挿入されていること(586_587insT)を特定しました。この挿入の結果、アミノ酸197番目でフレームシフトが起こり、12個の異常アミノ酸が追加されました。この突然変異により、高度に保存されているタンパク質のC末端半分の正常な翻訳が妨げられました。C4SLO細胞株における2番目の突然変異は特定されていません。
0.0005 スミス・レムリ・オピッツ症候群
DHCR7、HIS119LEU
スミス・レムリ・オピッツ症候群(SLOS)の特徴を持つ、血縁関係のない両親から生まれた男児において、Waterhamら(1998)は、デルタ-7-デヒドロコレステロール還元酵素遺伝子におけるhis119-to-leuおよびgly244-to-arg(602858.0006)変異の複合ヘテロ接合性を発見しました。この男児は出生時に低緊張で、小頭症、小顎症、頭蓋顔面異常、両手の軸後性多趾症、第2趾と第3趾の合指症、および重度の陰茎小帯下裂と思われる曖昧な性器が認められました。1歳時点で、重度の発達遅延が見られました。この患者におけるアミノ酸置換の最初のものは、母親から遺伝した356A-T転位によるものでした。2番目のものは、父親から遺伝した730G-A転移によるものでした。
0.0006 スミス・レムリ・オピッツ症候群
DHCR7、GLY244ARG
スミス・レムリ・オピッツ症候群(SLOS;270400)患者において複合ヘテロ接合状態で発見されたDHCR7遺伝子のgly244-to-arg(G244R)変異に関するWaterhamら(1998年)の研究については、602858.0005を参照のこと。
.0007 602858.0001 に移動.
0008 スミス・レムリ・オピッツ症候群
DHCR7、TRP248CYS
スミス・レムリ・オピッツ症候群(SLOS;270400)の乳児において、Waterham ら(1998)は、134bpの挿入(602858.0001)と744G-Tトランスバージョンによる複合ヘテロ接合性を発見し、その結果、DHCR7遺伝子において、トリプトファン248がシステインに置換しました。(Yu ら(2000年)は、IVS8-1G-C 変異(602858.0001)により、最後のエクソンであるエクソン9の異常なスプライシングが生じ、イントロン8の配列が134bp挿入され、その結果としてフレームシフトが起こり、早期に翻訳が停止すると指摘しています。)この男児は、子宮内発育遅延を伴う妊娠の後、正期産で生まれました。出生時、この男児は小頭症で、鼻先が広く鼻孔が前方に突出していること、軟口蓋裂、広い歯槽堤、小顎症、第2および第3足指の合指症、停留睾丸を伴う小さな陰茎など、複数の形態異常を示していました。3歳時、この男児は精神運動発達遅延、著しい発育不全、そして現在も経管栄養が必要な摂食障害がありました。SLO症候群は、血漿コレステロール値の低さと7-デヒドロコレステロール濃度の高さに基づいて生化学的に診断され、培養皮膚線維芽細胞における14-C-メバロン酸取り込みによって測定した7-DHCR活性の大幅な低下によって確認されました。
0.0009 スミス・レムリ・オピッツ症候群
DHCR7, THR93MET
スミス・レムリ・オピッツ症候群(SLOS)患者(270400)において、Fitzky ら(1998)は、DHCR7 遺伝子のヌクレオチド278におけるヘテロ接合性C-to-T 変異を同定し、その結果、Thr93がMetに置換(T93M)しました。De Brasi ら(1999年)は、T93M がイタリアの SLOS 患者9名において最も頻繁にみられる突然変異であり、18のアレリック中7つで発生していることを発見しました。
Witsch-Baumgartner ら(2001年)は、英国の22人のSLOS患者を対象とした研究で、44のアレリックのうち3つ(6.8%)にT93M変異を検出しました。この変異は、ドイツ/オーストリアの22人のSLOS患者、およびポーランドの15人の患者では検出されませんでした。
Scalco ら(2005年)は、ブラジル人SLOS患者14人を対象とした研究で、28のアレリックのうち9つ(32%)にT93M変異を検出しました。7人の患者では、T93M変異がIVS8-1G-C変異(602858.0001)との複合ヘテロ接合性で発見されました。
Witsch-Baumgartner ら(2008年)は、ヨーロッパのアレルのハプロタイプ分析とチンパンジーの Dhcr7 相同遺伝子との比較により、T93M 変異は約 6,000 年前に地中海東部で発生した可能性が高いと推定しました。
Kalb ら (2012) は、トルコの SLO 症候群患者 13 名から得た変異アレルの 26 個のうち 9 個 (36%) に T93M 変異を同定しました。 3 名の発端者はこの変異のホモ接合でした。 771 名の対照群には T93M 保因者は認められませんでした。 アレルの頻度は 420 人に 1 人以下と推定されています。
.0010 スミス・レムリ・オピッツ症候群
DHCR7、TRP151TER
Witsch-Baumgartner ら(2000年)は、重症のスミス・レムリ・オピッツ症候群(SLOS; 270400)(重症度スコアが50を超える)患者16人において、 32の疾患アレリックのうち5つが、DHCR7遺伝子のヌクレオチド453でG-to-A転移が起こり、W151X(W151からT152への置換)が生じていることを発見しました。
Loffler ら(2000年)は、W151Xの無効変異のホモ接合性により、重篤で致死的なSLOSを発症した2人の同胞について報告しています。1人は生後19日で死亡し、もう1人は妊娠20週で中絶されました。
Witsch-Baumgartner ら(2001年)は、英国、ドイツ、ポーランド、オーストリアの59人のSLOS患者を対象とした研究で、118のアレリックのうち19例(16.1%)にW151X変異を検出しました。東西の差異が認められました(ポーランドではアレリックの33.3%、ドイツとオーストリアでは18.2%、英国では2.3%)。DHCR7遺伝子のコーディング配列における8つの一塩基多型を用いたハプロタイプ解析により、W151X変異が3つの異なるハプロタイプで発生していることが分かりました。これらのハプロタイプは一塩基の位置のみが異なるため、同じ祖先ハプロタイプに由来する可能性があるため、著者らは、この変異が再発変異であるか、あるいは非常に古く、異なるハプロタイプに広がっている可能性のどちらであるかを区別できませんでした。
Ciara ら(2004年)は、ポーランド系民族の患者37人を対象とした研究で、68のアレリックのうち22個(32%)にW151X変異を発見しました。
ヨーロッパ人のアレリックのハプロタイプ分析とチンパンジーの Dhcr7 相同遺伝子との比較により、Witsch-Baumgartner ら (2008) は、W151X 変異は約 3,000 年前に北東ヨーロッパで出現したと推定しています。.
0011 スミス・レムリ・オピッツ症候群
DHCR7、VAL326LEU
スミス・レムリ・オピッツ症候群(SLOS;270400)の患者5人において、Fitzky ら(1998)はDHCR7遺伝子のヌクレオチド976でG-to-Tのトランスバージョンを発見し、val326-to-leu(V326L)のアミノ酸置換が生じていることが分かりました。これらの患者のうち3名では、V326L変異が複合ヘテロ接合状態で発見されましたが、他の2名の患者ではV326L変異は1つのアレリックのみで発見されました。 SSCP分析では、これらの2名の患者における2つ目の変異は発見されませんでした。
Yu ら(2000年)は、SLOS患者32人のスクリーニングで、V326L変異が64のアレリックのうち5つ(7.8%)を占めていることを発見し、これは彼らのコホートで観察された3番目に多い変異となりました。最も多く見られた変異の第1位と第2位は、それぞれIVS8-1G-C(602858.0001)とT93M(602858.0009)でした。
Witsch-Baumgartner ら(2001年)は、英国、ドイツ、ポーランド、オーストリアの59人のSLOS患者を対象とした研究で、118のアレリックのうち16(13.6%)にV326L変異を検出しました。東西の勾配が検出されました(ポーランドではアレリックの23.3%、ドイツとオーストリアでは18.2%、英国では2.3%)。DHCR7遺伝子のコーディング配列における8つの一塩基多型を用いたハプロタイプ解析により、5つのV326L染色体が同じハプロタイプを共有していることが分かり、創始者効果の証拠が示されました。
Ciaraら(2004年)は、ポーランド系患者37人を対象とした研究で、68のアレリックのうち19(28%)にV326L変異を発見しました。
0.0012 スミス・レムリ・オピッツ症候群
DHCR7, TRP37TER
スミス・レムリ・オピッツ症候群(SLOS; 270400)患者32人のうち、Yu ら(2000年)は、DHCR7遺伝子の1つのアレリックなエクソン4にG-to-A 変異が認められ、その結果、トリプトファン37がセリンに置換(W37X)している患者を1人発見しました。
0.0013 スミス・レムリ・オピッツ症候群
DHCR7, ARG352TRP
Witsch-Baumgartner ら(2001年)は、英国、ドイツ、ポーランド、オーストリアの59人のスミス・レムリ・オピッツ症候群(SLOS; 270400)患者を対象とした研究において、118のアレリックのうち7つ(5.9%)にR352W変異を検出しました。この変異には東西の勾配が認められました(ポーランド、アレリックの13.3%、ドイツ/オーストリア、6.8%、英国、なし)。
0.0014 スミス・レムリ・オピッツ症候群
DHCR7、ARG404CYS
Witsch-Baumgartner ら(2001年)は、英国、ドイツ、ポーランド、オーストリアの59人のスミス・レムリ・オピッツ症候群(SLOS)患者を対象とした研究において、DHCR7遺伝子の118のアレリックのうち5つ(4.2%)にR404C変異を検出しました(270400)。この変異には、西から東に向かう勾配が認められました(イギリスではアレリックの9.1%、ドイツとオーストリアでは2.3%、ポーランドでは認められず)。
0.0015 スミス・レムリ・オピッツ症候群
DHCR7、THR289ILE
Nowaczyk ら (1998) および Krakowiak ら (2000) が報告したスミス・レムリ・オピッツ症候群 (SLOS; 270400) の2人の兄弟において、Krakowiak らは DHCR7 遺伝子のヌクレオチド位置866でCからTへの変異を検出し、コドン289でトレオニンからイソロイシンへの変化 (T289I) が生じていることを発見しました。T289I 変異は、DHCR7 タンパク質の第 6 膜貫通領域と第 7 膜貫通領域の間に生じます。 この変異は、IVS8-1G-C 変異(602858.0001)との複合ヘテロ接合性で発見されました。 Nowaczyk ら(2001)は、これらの患者の両親および女性従姉妹にまで研究を拡大しました。 兄弟の父親は T289I ミスセンス変異を保有していました。従兄弟は兄弟と同様に、IVS8-1G-C/T289I遺伝子型を有していました。
0.0016 スミス・レムリ・オピッツ症候群
DHCR7、TYR280CYS
スミス・レムリ・オピッツ症候群(SLOS;270400)患者において、Prasad ら(2002)は、一般的なIVS8-1G-C(602858.0001)変異と、DHCR7におけるtyr280-to-cys(Y280C)変異の複合ヘテロ接合性を発見しました。影響が非常に軽微な女性乳児は、当初、摂食困難、発育不全、低緊張、軽度の発達遅延、口腔触覚嫌悪を呈していました。 軽微な顔面異常と両足の2~3指の合指症がありました。 血漿コレステロール値は2.88mmol/Lと境界線上の低値でした。 血漿7-デヒドロコレステロール値が200.0μmol/Lと上昇していたため、SLOSの臨床診断が確定しました。この患者における一般的な変異は既知のヌル変異であったため、Y280C変異は、臨床的表現型のより軽度の発現につながる、かなりの残存酵素活性と関連していると推定されます。
0.0017 スミス・レムリ・オピッツ症候群、軽度
DHCR7、MET1LEU
Langius ら (2003) は、スミス・レムリ・オピッツ症候群(SLOS; 270400)の非常に軽度の症状を持つ2人の兄弟において、一般的なIVS8-1G-Cヌル変異(602858.0001)と、メチオニンtRNA1(M1L)の翻訳開始に影響を与えるDHCR7遺伝子における新規変異の複合ヘテロ接合性を発見しました。15歳の兄は、比較的高い額と幅広い鼻梁、および比較的小さな顎を有すると記述されています。第2および第3足指の合指症および尿道下裂が認められました。10歳の弟も同様に、第2および第3足指の合指症および左第2指の彎指趾症を有していました。
0018 スミス・レムリ・オピッツ症候群、軽度
DHCR7、GLU448LYS
Langius ら (2003) は、スミス・レムリ・オピッツ症候群(SLOS; 270400)の非常に軽度の症状を持つ12歳の少女について報告しています。この症候群は、 一般的なIVS8-1G-Cヌル変異(602858.0001)と、DHCR7遺伝子における新規変異であるM1L(602858.0017)の複合ヘテロ接合性によるものであることが判明しました。さらに、1342G-A変異が認められ、グルタミン酸448がリジン(E448K)に置換していました。 頭囲は出生時に標準偏差の-3であり、12歳時には両側第2・第3足指合指症と高い額が認められました。
0.0019 スミス・レムリ・オピッツ症候群、軽度
DHCR7、PHE284LEU
Yu ら(2000年)は、非常に軽度のスミス・レムリ・オピッツ症候群(SLOS;270400)患者において、DHCR7 遺伝子における 2 つの変異(フェニルアラニン 284 がロイシンに置換する変異( エクソン8の膜貫通ドメインのすぐ外側にあるphe284からleu(F284L)と、エクソン9の膜貫通ドメインにあるより一般的なval326からleu(V326L; 602858.0011)です。Muellerら(2003)は、この患者の臨床的特徴について報告しています。発達面では、この子供は軽度の低緊張症に関連した粗大運動能力の軽度の遅れを示しました。認知能力、言語能力、運動能力はすべて、14パーセンタイルから18パーセンタイルの間の、その子の年齢の平均的な範囲を下回るものでした。多くのSLOS患者に見られるような、かんしゃく、運動の固定パターン、睡眠障害、光や音への過敏性は見られませんでした。この患者は生後12日目に、哺乳不良、腹部膨満、黄疸を呈して来院しました。大腸生検によりヒルシュスプルング病と診断されました。身体検査では、軽度の2,3指症が認められました。7-デヒドロコレステロール値は著しく上昇しており、コレステロール値は正常でした。生後3ヶ月でコレステロールの補給を行ったところ、コレステロール値は上昇し、7-デヒドロコレステロール値は低下しました。脳のMRIでは異常は認められませんでした。.
0020 スミス・レムリ・オピッツ症候群、軽症
DHCR7、MET1VAL
スミス・レムリ・オピッツ症候群(SLOS)の軽症患者(270400)において、Scalco ら(2005年)は1A-Gトランジションを発見し、その結果、DHCR7遺伝子の開始コドンにmet1-to-val(M1V)置換が生じました。Wassif ら(1998年)により機能的な DHCR7 タンパク質が生成されることが証明された met59 でのタンパク質合成が、軽度の表現型を説明できる可能性が示唆されました。
0.0021 スミス・レムリ・オピッツ症候群
DHCR7, ARG352GLN
日本人患者7人のスミス・レムリ・オピッツ症候群(SLOS; 270400)において、松本氏らは(2005年)、 その結果、高度に保存されたステロール感知領域である第8膜貫通ドメイン内に、アルギニン352からグルタミンへの置換(R352Q)が起こりました。2人の患者はR352Q変異のホモ接合型であり、変異型アレリックの14個中9個でこの変異が起こりました。ハプロタイプ分析により、創始者効果(founder effect)が示唆されました。
0.0022 スミス・レムリ・オピッツ症候群
DHCR7、IVS5DS、A-T、+3
スミス・レムリ・オピッツ症候群(SLOS;270400)の女児において、Koo ら(2010)は、DHCR7 遺伝子における2つの変異について複合ヘテロ接合性を確認しました。 C スプライス部位の変異(602858.0001)とイントロン5におけるA-Tトランスバージョン(IVS5+3A-T)が同定され、その結果、エクソン5のスキップ、フレームシフト、早期終結が起こります。患者の線維芽細胞のRT-PCR研究では、野生型バンドを含む3つのバンドが示され、IVS5+3A-T変異から野生型タンパク質がいくらか残存して産生されていることが示されました。しかし、患者の線維芽細胞はコレステロール欠損培地においてコレステロール合成の欠陥を示しました。患者は出生時に重度の障害があり、多くの器官系に影響を及ぼす複数の先天性異常がありましたが、出生後は予想よりも神経障害が軽度でした。生後7ヶ月で左右に転がり、生後11ヶ月で補助があれば立つことができ、細かい運動制御もいくらか可能になりました。4ヶ月齢の時点で血清7-デヒドロコレステロール値は上昇していましたが、その後正常範囲まで低下し、血清コレステロール値は正常でした。Koo ら(2010年)は、より重篤な表現型を示す患者と、より軽度の表現型を示す患者とを比較し、この患者には不一致が認められることを観察しました。この患者はより重篤な影響を受けていましたが、7-デヒドロコレステロール/コレステロール比はより軽度の影響を受ける患者で通常観察される値よりも低かったのです。Koo ら(2010年)は、胚発生の過程でコレステロールの必要性が非常に高いことを指摘しており、これがこの子供がこれほど多くの異常を持って生まれた理由を説明している可能性があります。出生後、IVS5+3A-T 変異によって残存する酵素活性と食事によるコレステロールの摂取により、ある程度の発達的獲得が可能になった可能性があります。







