目次
ATP7B遺伝子は、私たちの体内における銅の代謝と輸送に不可欠な役割を担っています。この遺伝子に変異が生じると、ウィルソン病という稀な遺伝性疾患を引き起こす可能性があります。本記事では、ATP7B遺伝子の機能、ウィルソン病との関連性、保因者検査の重要性について詳しく解説します。
ATP7B遺伝子の基本情報
ATP7B遺伝子(ATPase Copper Transporting Beta)は、13番染色体の長腕(13q14.3)に位置しています。この遺伝子は、P型ATPaseというタンパク質ファミリーに属する膜タンパク質をコードしており、主に肝臓や腎臓で発現しています。
ATP7B遺伝子の主な特徴
- 染色体位置:13q14.3
- 遺伝子サイズ:約80kb(キロベース)
- エクソン数:21個
- 主な発現部位:肝臓、腎臓、脳
- コードするタンパク質:銅輸送ATPase 2(1,465アミノ酸)
ATP7B遺伝子の機能
ATP7B遺伝子がコードするタンパク質は、P型ATPase銅輸送体と呼ばれる膜タンパク質で、主に肝細胞内のトランスゴルジネットワークに局在しています。このタンパク質は、生体にとって必須微量元素である銅の代謝において中心的な役割を果たしています。
ATP7Bタンパク質の主要な役割
- 銅イオンの細胞外排出:肝細胞内で過剰となった銅を細胞外(主に胆汁中)に排出し、最終的に便として体外に排泄する過程を担っています。これにより体内の銅バランスが維持されます。
- セルロプラスミンへの銅の取り込み:血清中の主要な銅結合タンパク質であるセルロプラスミンに銅を供給します。セルロプラスミンは体内の鉄代謝にも関与する重要なタンパク質です。
- 銅ホメオスタシスの維持:細胞内の銅濃度に応じて、その局在を変化させることで体内の銅濃度を適切なレベルに調節します。銅濃度が上昇すると、ATP7Bタンパク質はトランスゴルジネットワークから小胞体へと移動し、銅の排出を促進します。
- 銅依存性酵素の活性化:ATP7Bタンパク質は、様々な銅依存性酵素(リソチルオキシダーゼ、チロシナーゼなど)に銅を供給し、それらの活性化を助けます。
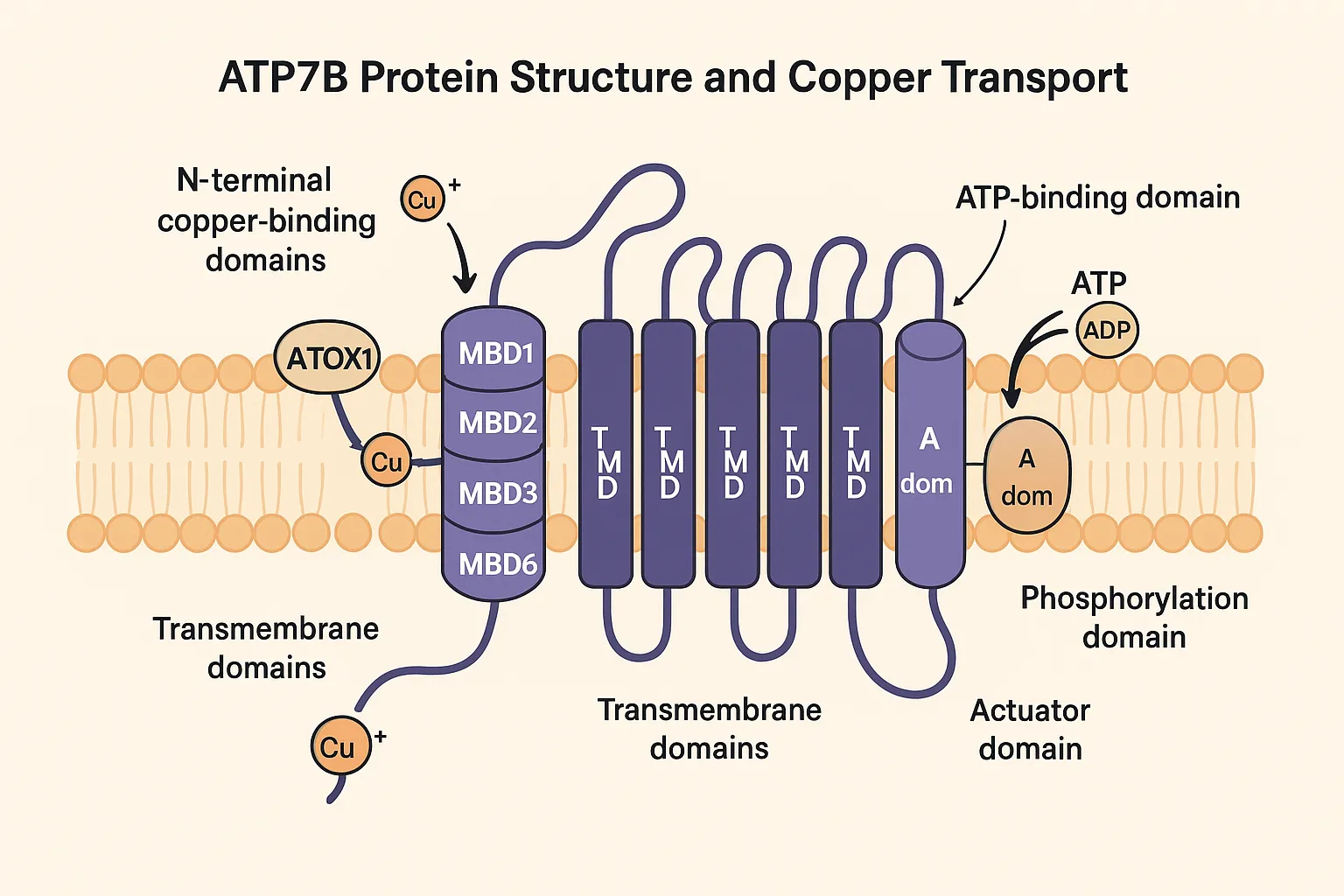
ATP7Bタンパク質の構造と機能ドメイン
ATP7B遺伝子がコードするタンパク質は1,465アミノ酸からなり、複数の機能ドメインを持つ複雑な構造をしています。
- N末端領域の銅結合ドメイン:N末端領域には6つの銅結合ドメイン(MBD1-6)があり、各ドメインにはCys-X-X-Cysモチーフが含まれています。これらのドメインが銅イオン(Cu+)と結合してタンパク質内部を輸送します。
- 膜貫通ドメイン:8つの膜貫通ドメイン(TMD)があり、これらが細胞膜やゴルジ膜を貫通して銅イオンのチャネルを形成します。特にTMD6にはCPC(システイン-プロリン-システイン)モチーフがあり、銅イオンの輸送に重要です。
- ATP結合ドメイン(N-ドメイン):ATPを結合し、加水分解することでエネルギーを得るドメインです。このエネルギーが銅イオンの能動輸送に使われます。
- リン酸化ドメイン(P-ドメイン):高度に保存されたDKTGTモチーフを含み、ATP加水分解によって生じたリン酸基が一時的に結合する部位です。
- アクチュエータードメイン(A-ドメイン):TGEモチーフを含み、脱リン酸化反応に関与します。
これらのドメインが協調して働くことにより、ATP7Bタンパク質は銅イオンの能動輸送を行います。まず、細胞質側でシャペロンタンパク質(ATOXl)から銅イオンを受け取り、ATP加水分解のエネルギーを使って膜の反対側(ゴルジ内腔や細胞外)へと輸送します。
ATP7Bと銅代謝サイクル
ATP7Bの銅輸送サイクルは以下のステップで進行します。
- ATOXlシャペロンからN末端の銅結合ドメインへの銅(Cu+)の受け渡し
- 銅イオンの膜貫通ドメイン内のCPCモチーフへの移動
- ATP結合と加水分解
- タンパク質のリン酸化と構造変化
- 銅イオンの膜の反対側への放出
- 脱リン酸化と初期状態への復帰
この一連のサイクルにより、銅イオンは濃度勾配に逆らって能動的に輸送されます。
ATP7Bタンパク質の機能は、銅の濃度に応じて巧妙に調節されています。銅濃度が低い状態では、主にトランスゴルジネットワークに局在してセルロプラスミンへの銅の取り込みを促進します。一方、銅濃度が上昇すると、小胞体を経由して細胞膜近傍へと移動し、銅の排出を促進します。この局在変化は、リン酸化・脱リン酸化などの翻訳後修飾によって制御されています。
ATP7B遺伝子とウィルソン病
ATP7B遺伝子の変異は、ウィルソン病(OMIM: 277900)と呼ばれる常染色体劣性遺伝性疾患を引き起こします。この疾患は、体内の銅代謝異常により、肝臓や脳などの組織に銅が過剰に蓄積することが特徴です。
ウィルソン病の主な症状
- 肝臓症状:肝炎、肝硬変、肝不全
- 神経症状:震え、筋強剛、構音障害、嚥下障害
- 精神症状:うつ、不安、性格変化
- 眼症状:カイザー・フライシャー輪(角膜の周辺部に現れる緑褐色の輪)
- その他:溶血性貧血、腎障害、骨・関節異常
ウィルソン病は適切な治療が行われないと重篤な肝障害や神経障害につながる可能性がありますが、早期発見と適切な治療により、症状の進行を抑えることができます。
ATP7B遺伝子の変異
現在までに、ATP7B遺伝子には700以上の病的変異(バリアント)が報告されています。これらの変異はエクソン全体にわたって分布しており、ミスセンス変異(55%)、ナンセンス変異(8%)、フレームシフト変異(17%)、スプライシング変異(9%)、プロモーター領域や非翻訳領域の変異(6%)、大きな欠失や挿入(5%)など様々なタイプが存在します。
変異の分布と好発部位
ATP7B遺伝子の変異は21のエクソン全体にわたって分布していますが、いくつかのホットスポットが存在します。
- エクソン8(銅結合ドメインと膜貫通領域の間)
- エクソン14(ATP結合ドメイン)
- エクソン16(ATP結合ドメイン)
- エクソン18(ATP結合ドメイン)
特にATP結合領域の変異は、タンパク質の機能に大きな影響を与えるため、重篤な症状を引き起こすことが多いとされています。
主なATP7B遺伝子変異
民族や地域によって頻度の高い変異が異なります。
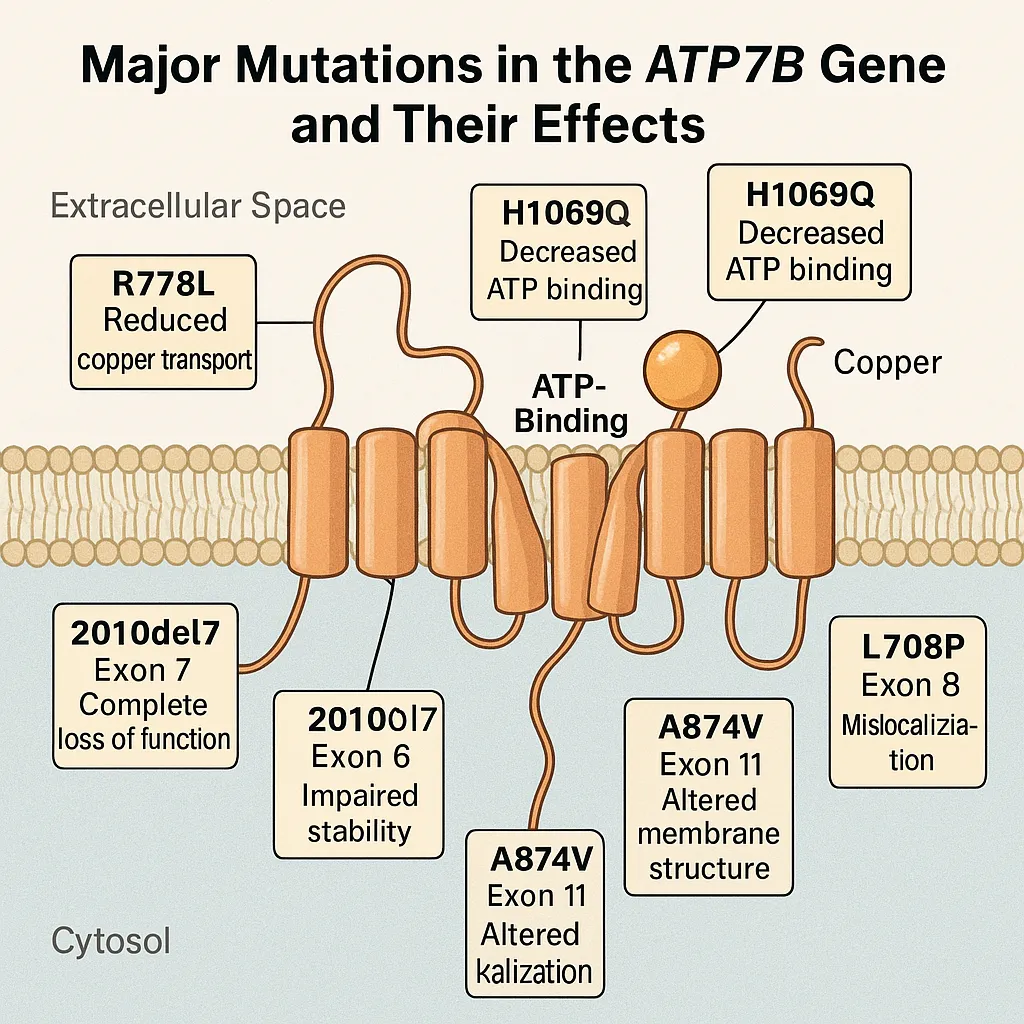
- H1069Q変異(エクソン14):ヨーロッパ系の患者で最も一般的(30-40%)で、ATP結合ドメインに位置し、ATP結合能を著しく低下させます。この変異を持つタンパク質は小胞体に異常に局在し、タンパク質の半減期も短縮されています。
- R778L変異(エクソン8):東アジア(日本、韓国、中国)の患者で最も一般的(30-40%)で、膜貫通領域に近いトランスメンブレンループに位置し、銅輸送効率を低下させます。韓国人では保因者頻度が0.6%とも報告されています。
- 2010del7(エクソン7):アイスランドで見られる7塩基の欠失で、フレームシフトを引き起こし、早期終止コドンを生じさせます。タンパク質のC末端半分全体が欠失するため、機能的に完全な喪失となります。
- M645R変異(エクソン6):スペイン人患者で高頻度(約55%)に見られ、6番目の銅結合ドメインと最初の膜貫通領域の間に位置しています。主に肝臓型のウィルソン病と関連しています。
- N1270S変異(エクソン18):コスタリカで高頻度(約61%)に見られ、ATPヒンジドメインに影響を与えます。韓国人では2番目に多い変異(約12%)としても報告されています。
- A874V変異(エクソン11):韓国人患者の約9%に見られる変異で、膜貫通領域の構造に影響を与えると考えられています。
- L708P変異(エクソン8):スペインのカナリア諸島で高頻度に見られ、神経学的症状を主体とする表現型と関連しています。
- 5’UTR -441/-427del15:サルデーニャ島の患者で特に多く見られる15塩基対の欠失で、転写活性を約75%低下させます。
変異タイプと表現型の関連
ATP7B遺伝子の変異はタンパク質の機能に様々な影響を与え、それが臨床像にも反映されます。
- タンパク質の細胞内局在に影響する変異:例えばH1069Q変異では、タンパク質が正しくトランスゴルジネットワークに局在できず、小胞体に留まってしまいます。これによりセルロプラスミンへの銅の取り込みが障害されます。
- 銅結合能に影響する変異:銅結合ドメインのCys-X-X-Cysモチーフに影響する変異は、銅イオンとの結合能を低下させます。
- ATP結合や加水分解に影響する変異:ATP結合ドメインの変異(H1069Q、N1270Sなど)は、エネルギー産生を阻害し、銅の能動輸送を妨げます。
- タンパク質の安定性に影響する変異:一部の変異はタンパク質の折りたたみや安定性に影響し、分解が促進されることで機能的なタンパク質量が減少します。
- スプライシングに影響する変異:スプライス部位の変異は異常なスプライシングを引き起こし、機能的なmRNAの生成を妨げます。例えばIVS4-1G-C変異は、エクソン5を欠失させ、54アミノ酸(最後の銅結合ドメインを含む)を失わせます。
遺伝型と表現型の複雑な関係
ATP7B遺伝子の変異と臨床症状の間には複雑な関係があります。以下のような特徴が観察されています。
- 同じ変異を持つ患者でも症状の発症時期や重症度が異なる場合がある
- 一般的に、タンパク質を完全に欠損させる変異(ナンセンス変異、フレームシフト変異など)は、部分的な機能を残すミスセンス変異よりも早期発症や重症化と関連する傾向がある
- 両アレルの変異の組み合わせ(複合ヘテロ接合性)が臨床像に影響を与える
- 環境因子(食事からの銅摂取量など)や修飾遺伝子が症状の発現に影響する可能性がある
変異の機能解析
ウィルソン病の変異の病的意義を評価するために、様々な機能解析が行われています。
- 酵母などのモデル生物を用いた銅輸送能の評価
- 培養細胞での変異タンパク質の局在解析
- in vitroでのATP加水分解活性の測定
- タンパク質の安定性や半減期の評価
- 銅依存性のタンパク質トラフィッキングの解析
これらの研究により、変異の病的意義や機能的影響についての理解が深まっています。例えば、一部の変異(E1173G変異など)は温度感受性を示し、低温(28℃)では機能が部分的に回復することが明らかになっています。このような知見は、将来的な治療法開発にも重要な手がかりを提供しています。
ATP7B遺伝子と保因者頻度
ATP7B遺伝子変異の保因者頻度は民族によって異なります。ウィルソン病は常染色体劣性(潜性)疾患であるため、両親から変異遺伝子を1つずつ受け継いだ場合(すなわち両アレルに変異がある場合)に発症します。一方、1つの変異アレルのみを持つ人は「保因者」となり、通常は症状を示しません。
| 対象人口 | 保因者頻度 | 検出率 | 検査後保因確率 | 残存リスク |
|---|---|---|---|---|
| 一般集団 | 1/87人 | 98% | 1/4,301人 | 1/1,496,748人 |
| コーカサス/ヨーロッパ系人口 | 1/42人 | 98% | 1/2,051人 | 1/344,568人 |
| アシュケナージ系ユダヤ人口 | 1/70人 | 98% | 1/3,451人 | 1/966,280人 |
日本人における保因者頻度の正確なデータは限られていますが、アジア人口全体では約1/90人程度と推定されています。日本人に多いR778L変異などの特定の変異の頻度も考慮すると、日本人の保因者検査は特に重要といえるでしょう。
ATP7B遺伝子検査の重要性
ATP7B遺伝子検査は以下のような場合に重要です。
- ウィルソン病の診断確定:臨床症状や生化学的検査でウィルソン病が疑われる場合
- 保因者検査:ウィルソン病の家族歴がある場合や、結婚前・妊娠前のカップルでリスク評価を行いたい場合
- 出生前診断:両親がともに保因者であることが判明している場合
- 新生児スクリーニング:早期発見・早期治療によって予後を改善できる可能性がある
保因者検査の意義
保因者検査は、自分がATP7B遺伝子変異の保因者であるかどうかを知ることができる検査です。特にウィルソン病の家族歴がある方やパートナーとともに検査を受けることで、将来の子どもがウィルソン病を発症するリスクを評価することができます。
両親がともに保因者である場合、子どもがウィルソン病を発症するリスクは25%、保因者になるリスクは50%、まったく変異を受け継がないリスクは25%となります。
ミネルバクリニックでは、拡大版保因者検査にてATP7B遺伝子を含む多数の遺伝子の保因者検査を提供しています。この検査では、将来のお子さんに影響を与える可能性のある遺伝子変異を事前に知ることができます。
ウィルソン病の診断と治療
ウィルソン病の診断は、臨床症状、生化学的検査(血清セルロプラスミン値、24時間尿中銅排泄量など)、画像検査、眼科検査(カイザー・フライシャー輪の有無)、肝生検、そしてATP7B遺伝子検査などを組み合わせて行われます。
ウィルソン病の治療法
- 銅キレート薬:D-ペニシラミン、トリエンチン、亜鉛製剤などを用いて体内の銅を除去する
- 食事療法:銅を多く含む食品(レバー、ナッツ類、チョコレートなど)の摂取制限
- 肝移植:重度の肝不全の場合
- 定期的なモニタリング:治療効果や病状の進行を評価するための定期検査
ウィルソン病は早期発見・早期治療が重要です。適切な治療により、多くの患者さんは症状の進行を抑えながら通常の生活を送ることができます。また、一度治療を開始したら、生涯にわたって継続する必要があります。
最新の研究と今後の展望
ATP7B遺伝子に関する研究は現在も進行中であり、以下のような分野で進展が見られています。
- 新規治療法の開発:より効果的で副作用の少ない銅キレート薬や、遺伝子療法などの開発
- バイオマーカーの探索:早期診断や治療効果のモニタリングに役立つ新たなバイオマーカーの同定
- 遺伝型と表現型の関連研究:特定の遺伝子変異と臨床症状やその重症度との関連性の解明
- モデル動物を用いた研究:ウィルソン病の病態メカニズムのさらなる理解や新規治療法の評価
これらの研究成果は、今後のウィルソン病の診断や治療の改善につながることが期待されています。
まとめ:ATP7B遺伝子とウィルソン病
ATP7B遺伝子は体内の銅代謝において重要な役割を果たしており、この遺伝子の変異はウィルソン病という稀な常染色体劣性遺伝性疾患を引き起こします。以下がポイントです。
- ATP7B遺伝子は肝臓での銅代謝に関わるタンパク質をコードしている
- 遺伝子変異により、体内に銅が過剰に蓄積し、肝臓や脳などに障害が生じる
- 症状は肝障害、神経障害、精神症状など多岐にわたる
- 適切な治療により、症状の進行を抑えることが可能
- 保因者検査は将来の子どもがウィルソン病を発症するリスクを評価するのに役立つ
ウィルソン病の家族歴がある方や、ご自身が保因者かどうか知りたい方は、遺伝カウンセリングをご検討ください。臨床遺伝専門医による適切な情報提供と心理的支援を受けることができます。
参考文献
- OMIM (Online Mendelian Inheritance in Man): ATP7B, 606882
- Wallace DF, Dooley JS. ATP7B genotype-phenotype correlations: Wilson’s disease manifestations and severity. Ann N Y Acad Sci. 2021.
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol. 2021.
- Coffey AJ, et al. A genetic study of Wilson’s disease in the United Kingdom. Brain. 2021.
- Gromadzka G, et al. Frequency of ATP7B gene mutations in Polish patients with Wilson disease. Neurol Neurochir Pol. 2022.

ミネルバクリニックでは、「未来のお子さまの健康を考えるすべての方へ」という想いのもと、東京都港区青山にて保因者検査を提供しています。遺伝性疾患のリスクを事前に把握し、より安心して妊娠・出産に臨めるよう、当院では世界最先端の特許技術を活用した高精度な検査を採用しています。これにより、幅広い遺伝性疾患のリスクを確認し、ご家族の将来に向けた適切な選択をサポートします。
保因者検査は唾液または口腔粘膜の採取で行えるため、採血は不要です。 検体の採取はご自宅で簡単に行え、検査の全過程がミネルバクリニックとのオンラインでのやり取りのみで完結します。全国どこからでもご利用いただけるため、遠方にお住まいの方でも安心して検査を受けられます。
まずは、保因者検査について詳しく知りたい方のために、遺伝専門医が分かりやすく説明いたします。ぜひ一度ご相談ください。カウンセリング料金は30分16500円です。
遺伝カウンセリングを予約する







