重要なお知らせ:Xp21隣接遺伝子欠失症候群は、新生児期から生命を脅かす重篤な症状を引き起こす可能性がある遺伝性疾患です。早期診断と適切な管理が生命予後を大きく左右します。
Xp21隣接遺伝子欠失症候群(Xp21 Contiguous Gene Deletion Syndrome)は、X染色体短腕のp21.2からp21.1領域に物理的に隣接して存在する複数の遺伝子が、染色体の一部欠失によって同時に失われることで発症する稀少なゲノム疾患です。この症候群は単一の疾患ではなく、欠失した個々の遺伝子の機能喪失に起因する、一見すると無関係に見える複数の臨床的特徴が一個人に複合的に現れる「隣接遺伝子症候群」の典型例です。
本記事では、この複雑な疾患について、最新の医学的知見に基づいて詳細に解説し、患者様やご家族が正確な理解を得るための包括的な情報を提供いたします。
疾患の概要
隣接遺伝子症候群としての特徴
Xp21隣接遺伝子欠失症候群は「隣接遺伝子症候群(contiguous gene syndrome)」の典型例です。これは、染色体の微細な欠失によって複数の遺伝子がまとめて影響を受け、それぞれの遺伝子機能喪失が個別に、あるいは複合的に作用して複雑な病態像を作り出す疾患群を指します。
類似疾患との比較:例えば、同様の機序で発症する22q11.2欠失症候群では、約30個の遺伝子が巻き込まれ、主要遺伝子TBX1の欠失により先天性心疾患が生じ、他の遺伝子欠失が精神発達遅延や免疫低下などの多様な合併症を引き起こします。22q11.2欠失症候群は最も一般的な微細欠失症候群であり、NIPTによる出生前スクリーニングの対象にもなっています。
欠失領域に含まれる主要遺伝子
Xp21.2-p21.1領域には、以下の重要な遺伝子が含まれています:
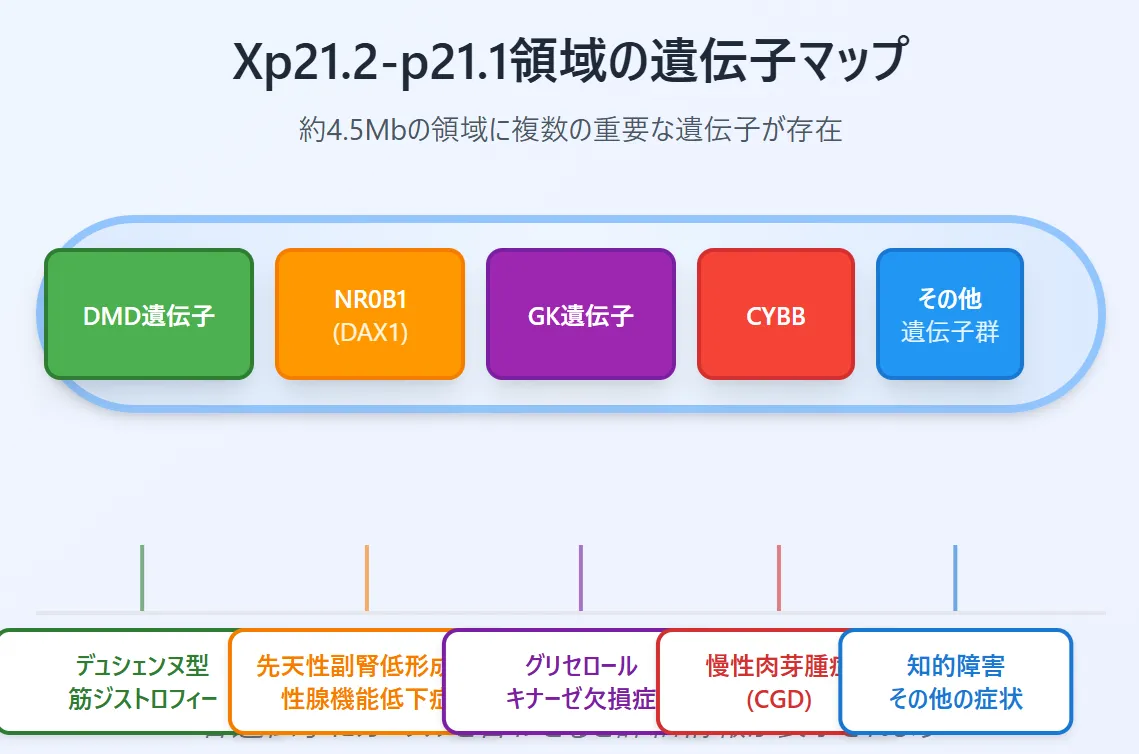
1. DMD遺伝子(Xp21.2)
- サイズ:約2.2メガベース(Mb)- ヒト最大の遺伝子
- 機能:ジストロフィンタンパク質をコード
- 役割:筋細胞膜の安定化、細胞骨格と細胞外基質の連結
- 関連疾患:デュシェンヌ型筋ジストロフィー(DMD)、ベッカー型筋ジストロフィー(BMD)
2. NR0B1(DAX1)遺伝子(Xp21.3~p21.2)
- 機能:核内受容体型の転写調節因子
- 役割:副腎や生殖腺の発生・機能維持
- 関連疾患:X連鎖性先天性副腎低形成症(AHC)、性腺機能低下症
3. GK遺伝子(Xp21.3)
- 機能:グリセロールキナーゼ酵素をコード
- 役割:グリセロールをグリセロール-3-リン酸に変換
- 関連疾患:グリセロールキナーゼ欠損症(GKD)
4. CYBB遺伝子(Xp21.1)
- 機能:NADPHオキシダーゼ複合体のgp91-phoxをコード
- 役割:好中球の殺菌能(活性酸素産生)
- 関連疾患:X連鎖性慢性肉芽腫症(CGD)
5. その他の重要遺伝子
- IL1RAPL1遺伝子:シナプス形成、X連鎖性知的障害との関連
- XK遺伝子:マクラウド症候群(赤血球異常、神経症状)
- OTC遺伝子:オルニチントランスカルバミラーゼ欠損症
- RPGR遺伝子:網膜色素変性症
なぜXp21領域は欠失しやすいのか
Xp21隣接遺伝子欠失症候群を深く理解するためには、「なぜこの領域が欠失しやすいのか」という疑問に答える必要があります。その答えは、この領域の特殊なゲノム構造にあります。特に、ヒト最大の遺伝子であるDMD遺伝子の巨大な構造と、そこに集まる特殊な配列が、この領域を「壊れやすい場所」にしています。
DMD遺伝子の巨大な構造
ヒト最大の遺伝子
DMD遺伝子は、ゲノム上で2.2メガベース(220万塩基対)以上という、ヒトの全遺伝子の中で最も大きなサイズを持っています。これは、ヒトゲノム全体の約0.1%に相当する広大な領域です。
この遺伝子は79個のエクソン(タンパク質を作るための設計図部分)と、78個のイントロン(設計図と設計図の間にある部分)から構成されています。驚くべきことに、遺伝子全体の99%以上がイントロンで占められています。つまり、実際にタンパク質を作るための情報は1%未満で、残りの99%以上は「間の部分」なのです。
例えば、イントロン44という部分だけで約250キロベース(25万塩基対)もの長さがあり、これはDMD遺伝子全体の約12%を占めています。このように、非常に広大な「間の領域」が存在することが、この遺伝子の大きな特徴です。
大きいことが壊れやすさにつながる理由
遺伝子が大きいということは、それだけエラーが起こる可能性のある「標的」が大きいということを意味します。DNAをコピーしたり修復したりする過程で、長い遺伝子ほどミスが起こりやすくなります。
実際に、DMDの発生率が男児出生3,500~5,000人に1人と比較的高く、その約3分の1が家族歴のない新しい変異であるという事実は、この遺伝子が構造的に壊れやすいことを示しています。
反復配列の集積
「動く遺伝子」の断片が集まっている
DMD遺伝子の広大なイントロン部分は、単なる空白ではありません。そこには、Alu配列という特殊なDNA配列が高密度に集まっています。Alu配列は「動く遺伝子」とも呼ばれる配列の断片で、ヒトゲノム中に100万コピー以上存在します。
これらの反復配列は、ゲノムの進化や多様性には役立つ一方で、ゲノムを不安定にする「火種」にもなります。DMD遺伝子の広大なイントロンは、これらの配列が蓄積する「受け皿」として機能してきました。その結果、蓄積した反復配列が遺伝子自身を壊してしまうという、皮肉な状況を生み出しています。
欠失が起こる仕組み
Xp21領域の欠失は、主にDNAが切れてしまった時の修復過程で起こるミスによって発生します。ここでは、2つの主要なメカニズムをご説明します。
修復ミスによる欠失
近年の最新技術を使った詳細な解析により、DMD遺伝子の欠失がどのように起こるかが明らかになってきました。多くの症例では、DNAが切れた時の「つなぎ直し」の過程でミスが起こることが原因とわかっています。
私たちの細胞には、切れたDNAをつなぎ直す仕組みがいくつかあります:
- 非相同末端結合(NHEJ):切れた部分を素早くつなぎ合わせる方法ですが、正確性は低く、つなぎ直す際に一部が失われることがあります。
- マイクロホモロジー媒介末端結合(MMEJ):短い似た配列を手がかりにつなぐ方法で、やはり欠失が起こりやすい仕組みです。
これらの修復方法は迅速ですが不正確なため、つなぎ直す過程で本来あるべきDNAの一部が失われてしまうことがあります。特に、広大なイントロン領域では、このような修復ミスが起こりやすいと考えられています。
Alu配列による「間違った組み換え」
もう一つの重要な仕組みは、前述したAlu配列が原因となる「間違った組み換え」です。これは非アレル相同組換え(NAHR)と呼ばれる現象です。
Alu配列は互いに非常によく似ています。そのため、細胞分裂の過程で、本来組み合わせるべき正しい位置のAlu配列ではなく、近くにある別のAlu配列と間違って組み合わせてしまうことがあります。この「間違ったペアリング」の後に組み換えが起こると、2つのAlu配列に挟まれたDNA領域が丸ごと失われてしまいます。
DMD遺伝子のイントロンには、Alu配列が高密度に存在しています。そのため、この「間違った組み換え」が起こる確率が非常に高くなっています。実際に、様々な遺伝性疾患でAlu配列を介した欠失が報告されており、Xp21領域の脆弱性の大きな原因となっていると考えられています。
特定の場所に集中する切断点
欠失が起こる場所(ブレークポイント)を詳しく調べると、特定の領域に集中している傾向が見られます。例えば、イントロン44では欠失の開始点の約25%が特定の場所に集まっているという報告があります。
この理由として、以下のような要因が考えられています:
- 特定のDNA配列が特殊な形(ヘアピンや十字架のような構造)を作りやすく、これがDNAのコピーを妨げたり、切断されやすくなったりする
- DNAのコピーが始まる場所で異常なコピーが起こり、それがゲノムの不安定性につながる
Xp21領域が欠失しやすい理由は、単一の原因ではありません。DMD遺伝子の巨大なサイズ、イントロンに蓄積した反復配列、DNA修復時のエラー、そして間違った組み換えといった複数の要因が複雑に絡み合って、この領域を「壊れやすい場所」にしています。このような分子レベルの理解は、将来的な予防法や治療法の開発にもつながる重要な知見です。
主要構成疾患の詳細
1. デュシェンヌ型筋ジストロフィー(DMD)
疾患の特徴
DMDは、通常2~5歳頃に発症する進行性の筋力低下を特徴とします。患児は歩行開始の遅れや、床からの立ち上がりに手を使う「Gowers(ガワーズ)徴候」を示します。
主な症状
- 発症年齢:2~5歳頃
- 初期症状:転倒しやすい、階段昇降困難、Gowers徴候
- 特徴的所見:ふくらはぎの仮性肥大、血清CK著明高値(正常の50~数百倍)
- 進行:12歳頃までに歩行能力喪失、車椅子依存
- 合併症:拡張型心筋症(18歳以降ほぼ全例)、呼吸不全
- 予後:20歳代で呼吸不全や心不全により死亡する例が多い
病態メカニズム:ジストロフィンタンパク質の完全な欠損により、筋細胞の骨格と細胞外の支持組織を繋ぐアンカー機能が失われます。この保護機能の喪失により筋細胞膜が脆弱化し、繰り返される筋収縮によって膜に微細な断裂が生じ、細胞内にカルシウムイオンが過剰に流入することで、最終的に筋細胞は壊死に至ります。この破壊と再生のサイクルが繰り返されるうちに再生能力が枯渇し、筋組織が脂肪や線維組織に置き換わっていきます。
2. X連鎖性先天性副腎低形成症(AHC)
生命を脅かす副腎クリーゼ
AHCの最も重篤な症状は、新生児期から乳児期にかけて発症する原発性副腎不全です。副腎皮質は、生命維持に不可欠なホルモン(コルチゾール、アルドステロン)を産生しますが、AHCではこれらのホルモンを十分に産生できません。
主な症状
- 発症時期:生後数週間から数ヶ月以内
- 初期症状:嘔吐、哺乳不良、体重増加不良、脱水
- 検査所見:低ナトリウム血症、高カリウム血症、低血糖
- 特徴的所見:皮膚の色素沈着(ACTH過剰分泌による)
- 緊急性:迅速な診断とホルモン補充療法がなければ生命を脅かす
- 思春期以降:性腺機能低下症(思春期遅発、男性不妊)
発症頻度:約60%は生後1か月以内に、40%は乳幼児期(1歳~9歳)までに副腎不全症状で発見されます。治療せずに経過すると致死的となり得ます。副腎不全を乗り越えても、思春期に第二次性徴の遅れ(ゴナドトロピン分泌不全)が明らかになる場合があります。
3. グリセロールキナーゼ欠損症(GKD)
疾患の特徴
グリセロールキナーゼ酵素の欠損により、脂肪組織で中性脂肪が分解されて生じるグリセロールを適切に代謝できなくなり、血中および尿中にグリセロールが蓄積します。
臨床型
- 乳児型(重症型):重篤な発達遅延、筋緊張低下、嘔吐、代謝性アシドーシス、低血糖発作。多くはXp21隣接遺伝子欠失症候群の一部として発症
- 若年型(症候性):症状は乳児型に似るが、より軽度で可変的
- 成人型(無症候性):臨床症状を全く伴わず、健康診断などで偶然発見されることが多い
診断の落とし穴:一般的な臨床検査室での中性脂肪(トリグリセリド)測定は、検体中のトリグリセリドをリパーゼで分解し、遊離したグリセロールの量を測定することで間接的に行われます。GKD患者では、血中に蓄積した遊離グリセロールがこの測定系で検出されるため、実際の中性脂肪値は正常であるにもかかわらず、見かけ上、著しい高中性脂肪血症と報告されることがあります。血清の外観に濁りを欠くことが示唆的です。
4. 慢性肉芽腫症(CGD)
疾患の特徴
CYBB遺伝子の欠失により、好中球などの食細胞が細菌や真菌を殺菌する能力(呼吸バースト)を失います。その結果、生命を脅かす重篤な感染症を反復する免疫不全症を呈します。
主な症状
- 発症時期:生後数週~数ヶ月
- 主症状:重篤な細菌・真菌感染症の反復(敗血症、肺炎、肝膿瘍など)
- 原因菌:カタラーゼ陽性菌(ブドウ球菌、アスペルギルスなど)
- 特徴:肉芽腫形成、抗生剤治療に反応しにくい
5. その他の合併疾患
- オルニチントランスカルバミラーゼ欠損症(OTCD):新生児期から高アンモニア血症による哺乳不良・嘔吐、昏睡。重症型では男児新生児期に発症し、治療が遅れると死亡することもある
- 知的障害:IL1RAPL1遺伝子の欠失による非症候性のX連鎖知的障害
- 網膜色素変性症:非常に広範な欠失でRPGR遺伝子まで含まれると、思春期以降に夜盲や視野狭窄
- マクラウド症候群:XK遺伝子の欠失により、赤血球形態異常(有棘赤血球症)、中年以降に舞踏病様の神経症状や心筋症
診断アプローチ
臨床的疑いのポイント
本症候群の診断で最も重要なのは、異なる臓器系にまたがる一見無関係な症状の組み合わせに気づくことです。
- 新生児期の副腎不全 + 著明なCK高値
- 副腎不全 + 筋力低下 + 反復感染
- 偽性高トリグリセリド血症 + 筋症状
- 高アンモニア血症 + 副腎不全
生化学的検査
- 血清クレアチンキナーゼ(CK):DMDでは正常上限の数十倍から100倍以上の著明な高値
- 血中・尿中グリセロール:GKDの証明
- ホルモン検査:コルチゾール低値、ACTH著明高値、血漿レニン活性高値、アルドステロン低値
- 好中球機能検査:NBT還元能試験、ジヒドロロダミン蛍光染色検査(CGD診断)
遺伝学的確定診断
染色体マイクロアレイ解析(CMA)- ゴールドスタンダード
CMAは、本症候群の確定診断における現在のゴールドスタンダードです。ゲノム全体のコピー数変化(CNV)を網羅的に、かつ高解像度で検出し、Xp21領域の欠失の有無、サイズ、および欠失に含まれる遺伝子群を一度の検査で明らかにすることが可能です。
MLPA法
特定の遺伝子(DMD、NR0B1、GKなど)のエクソン単位の欠失や重複を検出することに特化した検査法です。DMDが強く疑われる場合の第一選択検査として用いられることが多く、比較的安価かつ迅速に結果が得られます。
治療と管理
集学的治療チームの重要性
Xp21隣接遺伝子欠失症候群は、内分泌、神経筋、心臓、代謝、発達など、複数の臓器システムに生涯にわたる影響を及ぼします。そのため、患者の成長と病状の進行に合わせて、多岐にわたる専門家が連携する集学的治療チームによる包括的なケアが不可欠です。
各疾患に対する治療
ミネルバクリニックの特徴
- • COATE法:最新の次世代NIPT技術により自院採用検査のなかで最高精度を実現(微細欠失症候群従来検査の陽性的中率70%台➡99.9%)
- • 臨床遺伝専門医常駐:専門医による遺伝カウンセリング
- • オンライン対応:全国どこからでも受検可能
- • 24時間サポート:陽性時の手厚いフォロー体制
- • 国内最大12か所13疾患の微小欠失症候群を検査可能(2025年8月2日ミネルバクリニック調べ)
- • 国内唯一父親の高齢化により精子に生じる突然変異による疾患を56遺伝子検査可能(2025年8月2日ミネルバクリニック調べ)
- • 陽性時の確定検査を自院で可能:NIPT検査から陽性時の確定検査までワンストップで対応いたします
よくある質問(FAQ)
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
まとめ
Xp21隣接遺伝子欠失症候群は、X染色体p21領域の特異なゲノム構造に起因する脆弱性によって生じる、複数の疾患を合併する稀少なゲノム疾患です。その病態は、欠失領域に含まれるDMD、NR0B1、GKをはじめとする複数の遺伝子の機能喪失が加算的に作用した結果として理解されます。
重要なポイント:
- 早期診断の重要性:新生児期の副腎クリーゼや重症感染症は生命を脅かすため、迅速な診断と治療が必須
- 症候群的視点:異なる臓器系の症状の組み合わせに気づくことが診断の鍵
- 集学的アプローチ:多職種連携による包括的な長期管理が不可欠
- 遺伝カウンセリング:家族計画に関する適切な情報提供と心理社会的サポート
- 治療の進歩:DMDに対する遺伝子治療など、革新的な治療法の実用化により予後改善が期待される
この疾患は症状が多様であるからこそ、個々の患者様に応じた個別化されたアプローチが最も重要となります。最新の医学的知見に基づいた適切な管理により、患者様とご家族がより良い生活の質を維持できるよう、継続的なサポートを提供することが我々医療従事者の役割です。
参考文献
- Orphanet: Xp21 deletion syndrome. www.orpha.net/en/disease/detail/261476
- Rathnasiri et al. (2021). A rare co-occurrence of duchenne muscular dystrophy, congenital adrenal hypoplasia and glycerol kinase deficiency due to Xp21 contiguous gene deletion syndrome: case report. BMC Endocrine Disorders.
- Frontiers in Genetics (2022). Chronic granulomatous disease associated with Duchenne muscular dystrophy caused by Xp21.1 contiguous gene deletion syndrome: Case report and literature review.
- GeneReviews: Dystrophinopathies. NCBI Bookshelf.
- GeneReviews: NR0B1-Related Adrenal Hypoplasia Congenita. NCBI Bookshelf.
- Journal of Clinical Research in Pediatric Endocrinology (2025). Xp21 Contiguous Gene Deletion Syndrome: Diagnosis, Treatment, and a Review of the Literature on a Rare Genetic Disorder.
本記事の内容は医学的情報の提供を目的としており、特定の医学的アドバイスを意図するものではありません。症状や治療に関する具体的なご相談は、必ず専門医にご相談ください。







