目次

📍 クイックナビゲーション
13q14微小欠失症候群(第13染色体長腕部分モノソミー)は、第13染色体の長腕における特異的なゲノム領域の物理的喪失によって引き起こされる、極めて稀で複雑な先天性の染色体異常症候群です。
本疾患は、OrphanetやOMIMなどの主要な希少疾患データベースにおいて独立した疾患概念として確立されており、出生10,000人から40,000人に1人の割合で関連する染色体異常が発生していると推定されています。
1. 13q14微小欠失症候群の中核となる「三徴」
本疾患の臨床的な特徴は患者様によって多岐にわたりますが、診断の要となるのは以下の3つの主要な症状(三徴)です。
- 小児期の網膜芽細胞腫(Retinoblastoma)の極めて高い発症リスク
- 多様な程度の精神運動発達遅滞および知的障害(ID)
- 特徴的な頭蓋顔面形態異常(顔貌の特徴)
そもそもこの疾患は、網膜芽細胞腫の発症要因を遺伝学的に探求する過程で同定されました。網膜芽細胞腫全体の約5%〜15%は、遺伝子内の微小な点突然変異ではなく、RB1遺伝子を含む広範な欠失(13q-)によって引き起こされることが明らかになっています。
2. 分子遺伝学的基盤と発症のメカニズム(遺伝形式)
第13染色体は全体的に遺伝子密度が著しく低いという特性があります。広範な欠失があっても胎生致死とならずに出生に至る症例が存在するのはこのためです。しかし、13q14領域には細胞周期の制御を行う「RB1遺伝子」や、中枢神経系の発達に関与する重要な遺伝子クラスターが集中しているため、この領域が欠失すると重篤な臨床症状を惹起します。
🔍 関連してよく読まれている記事
対になる2本の染色体のうち、片方の遺伝子に変異があるだけで症状が現れる遺伝形式を常染色体優性(顕性:けんせい)と呼びます。両方に変異が必要な場合は常染色体劣性(潜性:せんせい)と呼びます。13q14微小欠失症候群は、変異が片方のアレルに起こるだけで発症するため、常染色体優性(顕性)の形式をとります。
本症候群の大部分は、親の生殖細胞(精子または卵子)の形成過程や初期胚発生において偶発的に生じる新生突然変異(de novo mutation)による散発例です。そのため、ご両親の染色体が正常であれば、兄弟姉妹における再発リスクは極めて低いと考えられます。
ただし例外として、ご両親のいずれかが第13染色体を含む「均衡型転座」や「複雑な染色体再構成(CCR)」の無症候性保因者である場合、生殖細胞の分裂時の不分離によって児に不均衡型の欠失が生じることがあります。再発リスクを正確に評価するためには、ご両親の核型分析およびFISH検査が不可欠です。
3. 欠失サイズによる症状の違い(遺伝子型-表現型相関)
症状の重症度や多様性は、欠失領域の「物理的なサイズ(メガベース:Mb)」と、そこに巻き込まれた「個々の遺伝子の種類」に強く依存します。欠失サイズは患者様ごとに異なり、共通のブレークポイント(切断点)は存在しません。
| 欠失の分類・サイズ | 特徴的な成長・発達の表現型 | 特徴的な頭蓋顔面形態異常 |
|---|---|---|
| 小規模欠失(Small) 6 Mb未満 (13q14領域内) |
大頭症、高身長、肥満などの過成長の兆候。運動・言語の発達遅滞は認められるが比較的軽度。 | 高く広い前額部、広い鼻尖、薄い上唇。 |
| 中規模欠失(Medium) 6 Mb〜20 Mb |
低身長および小頭症への移行。軽度から中等度の精神運動遅滞。 | 高い前額部、奥まった眼、短い鼻(幼少期)、小さな上唇、高い頻度での巻き毛。 |
| 大規模欠失(Large) 20 Mb以上 (13q12〜q31.2領域) |
著明な低身長、小頭症。軽度から最重度の精神運動遅滞。重度の筋緊張低下、難治性の便秘、深刻な摂食障害。 | 年齢とともに変化する顔貌(丸顔→長い顔貌)。長い人中、口角の下がった口元、眼距開離、耳介低位、小顎症。 |
マイクロアレイ染色体検査(array-CGH)の普及により、特定の遺伝子と症状の結びつきが解明されてきました。
- NBEA(Neurobeachin)遺伝子:知的障害や自閉症スペクトラム障害、発達遅滞の最も有力な候補遺伝子です。
- EDNRB遺伝子:ヒルシュスプルング病(腸管無神経節症)に類似した重篤な便秘、摂食障害、難聴に寄与しています。
- MED4遺伝子:この遺伝子が欠失から免れて保持されている場合、網膜芽細胞腫の表現型が軽症化する傾向があります。
4. 全身性合併症と多系統への影響
13q14欠失症候群は、眼科学的な問題にとどまらず、VACTERL連合(椎体異常、鎖肛、心疾患、気管食道瘻、腎奇形、四肢異常)に類似した広範な多系統の先天異常を合併する複合的な症候群です。
中枢神経系・筋骨格系の異常:出生直後から著明な「軸性筋緊張低下」を呈し、運動発達の遅れや新生児期の摂食困難の原因となります。MRI画像診断では、脳梁の無形成/低形成、小脳低形成などが頻繁に確認されます。骨格奇形として、第5指の斜指症や短指症なども観察されます。
心血管系・消化器系の異常:心室中隔欠損症(VSD)などの先天性心疾患の合併は生命予後に直結します。消化器系では、腸回転異常症に伴う腸閉塞、鎖肛など、早期の外科的介入を要する重篤な奇形を伴うことがあります。
顔貌の特徴に加え、眼球自体の構造的異常(網脈絡膜コロボーマ、白内障、小眼球症、斜視、視神経低形成など)も、網膜芽細胞腫とは独立して多発する点に注意が必要です。
5. 網膜芽細胞腫の病態メカニズムと厳格な検査プロトコル
本疾患の臨床管理において、生命および視覚機能の予後を決定づける最大の独立因子は網膜芽細胞腫の発症です。
RB1遺伝子(13q14.2に局在)は細胞周期を制御するがん抑制遺伝子です。患者様は出生時からすべての細胞に「ファースト・ヒット(一次変異)」を有しており、網膜の発生過程で残りの正常なアレルに「セカンド・ヒット(二次変異)」が生じることで悪性腫瘍が形成されます。
【極めて重要な注意点】 血液検査などで確認される欠失が「モザイク型(一部の細胞のみ欠失)」であっても、非モザイク型であっても、網膜芽細胞腫の発症リスクに統計的な有意差はありません。非浸透例はわずか7%に過ぎず、残り約93%が高い確率で発症に至ります。13q14欠失が診断された瞬間から、無条件で緊急かつ継続的な眼科的評価が必須です。
眼科的スクリーニングの標準ガイドラインと「三側性」の監視
米国眼科腫瘍病理学会(AAOOP)のガイドラインに基づき、特に腫瘍発生のリスクが高い乳児期は、無症状であっても全身麻酔下(EUA)での広角眼底検査を行い、微小な病変を見逃さないことが標準です。
- 生後0週〜8週: 2〜4週間ごとに非鎮静下での眼科検査。
- 生後8週〜12ヶ月: 毎月(1ヶ月ごと)全身麻酔下検査(EUA)。
- 12ヶ月〜36ヶ月: 2〜3ヶ月ごとの全身麻酔下検査。
- 7歳以降: 生涯にわたる定期検診。
さらに、眼球内だけでなく頭蓋内(主に松果体など)に原発性の腫瘍を発症する三側性網膜芽細胞腫(Trilateral Retinoblastoma: TRB)のリスクがあります。これは予後が極めて不良であるため、診断時のベースラインMRIと、4歳に達するまでの6ヶ月ごとの定期的なMRIスクリーニングが不可欠です。
最新治療と13q欠失患者特有の「毒性」への配慮
現在、眼球摘出や外照射を極力回避し、眼動脈注入化学療法(OAC)などを用いて視力を温存する治療が集学的に行われています。MSKCCの報告では、対象眼球の83%で2年間の眼球温存が達成されるなど高い治癒率を誇ります。
しかし、13q欠失を有する患者様は、通常のRB1遺伝子変異のみの患者様と比較して、化学療法に対する全身性の血液毒性を著しく生じやすいという事実があります。局所デリバリーであるはずのOACでも重篤な骨髄抑制(好中球減少症)を引き起こすため、治療後7〜14日の厳密な全血球計算(CBC)モニタリングと、過度な骨髄抑制を回避するための迅速な用量調整が小児腫瘍医との連携のもとで必須となります。
6. 成人における体細胞突然変異としての13q14欠失(CLLの予後)
ここまで小児期の先天性症候群について解説しましたが、このゲノム領域の病態を深く理解する上で、成人領域の慢性リンパ性白血病(CLL)における「後天的な体細胞突然変異」としての13q14欠失の役割を知ることも極めて重要です。
CLL患者の50%〜60%において、腫瘍細胞内に後天的な13q14領域の欠失が同定されます。この発がんドライバーはRB1遺伝子ではなく、同領域に位置する腫瘍抑制遺伝子座(DLEU2)およびそれに内包されるマイクロRNAの喪失です。これにより、抗アポトーシスタンパク質(BCL2)が過剰発現し、異常なB細胞が不死化して蓄積します。
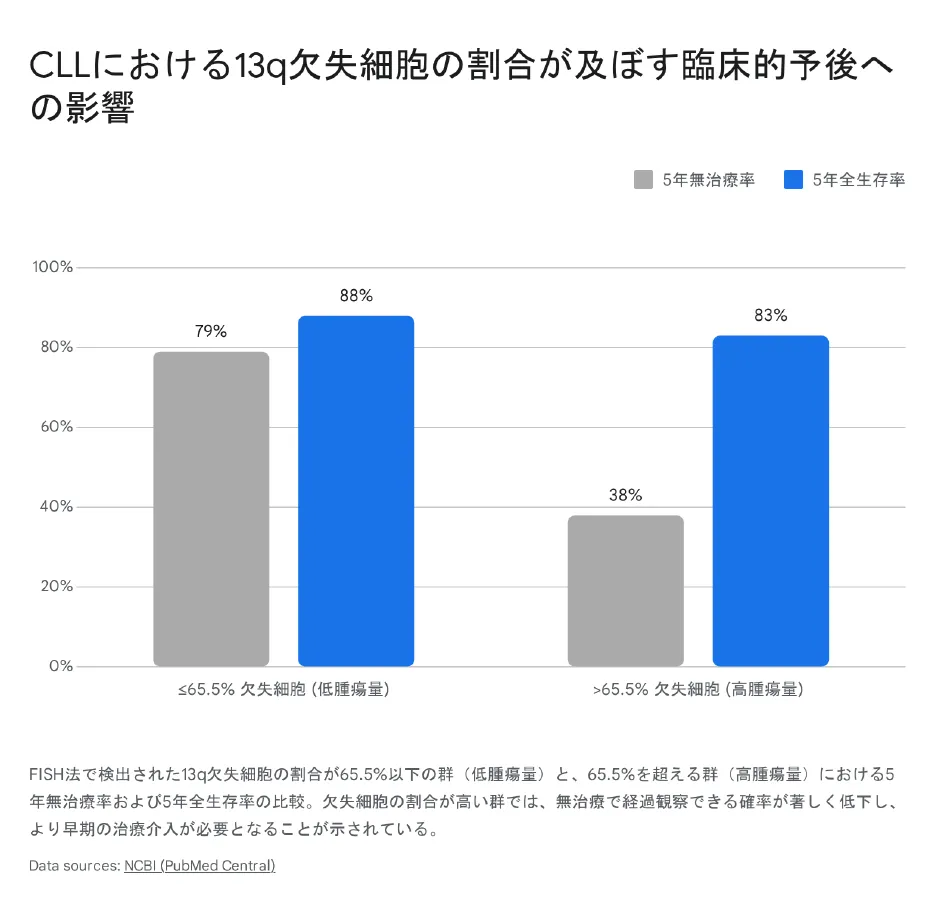
画像:FISH法で検出されたCLLにおける13q欠失細胞の割合(クローン量)が及ぼす臨床的予後への影響
一般的に、CLLにおいて13q14欠失が「単独の染色体異常」として検出される場合は最も予後良好なマーカーとされます。しかし近年の研究で、単なる欠失の有無ではなく「欠失を有する腫瘍細胞の割合(腫瘍量)」が予後をダイナミックに左右することが判明しました。
上記のデータが示す通り、13q欠失細胞の割合が65.5%以下の患者群は5年無治療率が79%と高く安定した経過をたどりますが、65.5%を超える高腫瘍量患者群では5年無治療率が38%へ急減し、より早期の治療介入が必要となります。
小児の先天性欠失が「物理的サイズと遺伝子の種類」で症状が決まるのに対し、成人の後天性欠失は「細胞集団が占有する割合」が疾患の攻撃性を決定するという、生物学的な挙動の鮮やかな対比がここに存在します。
7. 寿命・長期生存について:予後と集学的医療体制の構築
13q14微小欠失症候群の患者様は、適切な早期発見と治療が行われれば、長期生存は十分に可能です。診断後20年を経過した時点での追跡調査において、患者様の90%以上が生存しており、過去の文献では64歳まで生存した症例も報告されているなど、寿命に関する前向きなデータも存在します。
しかし、全身の組織で「ファースト・ヒット」を有しているため、成長後に骨肉腫や軟部肉腫などの二次性悪性腫瘍(SMN)を発症するリスクが生涯にわたり高いという宿命を背負っています。
特に放射線治療を受けた部位での発生頻度が高いため、最新治療では放射線の使用を極力回避します。小児期を脱した後も、生涯にわたるがんサーベイランスと予防的介入(厳格な紫外線対策、皮膚や骨の定期的観察)が不可欠です。

よくある質問(FAQ)
関連記事
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
参考文献
- Genetic and Rare Diseases Information Center (GARD) – Chromosome 13q14 deletion syndrome [GARD]
- Orphanet – VACTERL/VATER association [Orphanet]
- National Center for Biotechnology Information (NCBI) – Chromosome 13q14 deletion syndrome [NCBI MedGen]
- American Academy of Ophthalmology – First U.S. Guidelines for Retinoblastoma Screening [AAO]
- PubMed Central – 13q Deletion Syndrome Involving RB1: Characterization of a New Minimal Critical Region for Psychomotor Delay [PMC]
- PubMed Central – 13q14 Deletion and Its Effect on Prognosis of Chronic Lymphocytic Leukemia [PMC]






