目次
10p14-p13欠失症候群(DiGeorge症候群2型)の原因と症状

📍 クイックナビゲーション
10p14-p13欠失症候群は、ヒトの第10染色体短腕(p腕)の一部が欠落することによって引き起こされる、極めて稀で複雑な先天性染色体異常症です。この病気は、単一の症状に留まらず、心疾患、免疫不全、難聴、発達遅滞など多岐にわたる症状を合併する特徴があります。
この記事では、臨床遺伝専門医の視点から、本疾患の根本的な原因となる遺伝子のメカニズム、全身に現れる症状の理由、類似疾患である「22q11.2欠失症候群(DiGeorge症候群1型)」との決定的な違い、そして生涯を見据えた治療方針までを網羅的に解説します。
1. 第10染色体短腕遠位欠失とは?病態の全体像
第10染色体短腕遠位欠失(Distal monosomy 10p)は、染色体の末端領域における遺伝物質の不可逆的な喪失が原因です。かつては細胞遺伝学的なGバンド分染法で大まかに特定されていましたが、症状の多様性が極めて大きく、単一の疾患として捉えることが長らく困難でした。
しかし、近年の染色体マイクロアレイ検査(CMA)や次世代シーケンシング(NGS)技術の飛躍的な進歩により、この疾患の実態が「隣接遺伝子欠失症候群」であることが解明されました。
染色体上の物理的に隣り合っている「複数の独立した重要な遺伝子」が、まとまってすっぽりと抜け落ちてしまう現象です。一つの遺伝子の異常ではなく、複数の遺伝子が同時に失われるため、全く異なる複数の臓器(心臓、腎臓、脳など)に同時に重篤な症状が現れるのが特徴です。
本疾患の重症度は、欠失の切断点(ブレイクポイント)がテロメア側(末端の10p15領域)からセントロメア側(中心に近い10p14、さらには10p13領域)へと、どの程度深く及んでいるかに極めて強く依存します。欠失の範囲が広がるほど、巻き込まれるマスターレギュレーター遺伝子(他の多数の遺伝子の働きを指揮する重要な遺伝子)が増え、症状は累積的に重症化していきます。
2. 欠失領域と責任遺伝子の詳細メカニズム
10p欠失に伴う複雑な症状を理解するためには、染色体上のどの位置に、どのような役割を持つ遺伝子が存在しているかを紐解く必要があります。以下の図は、第10染色体短腕(10p)の遺伝子マッピングと、関連する症候群の相関構造を示したものです。
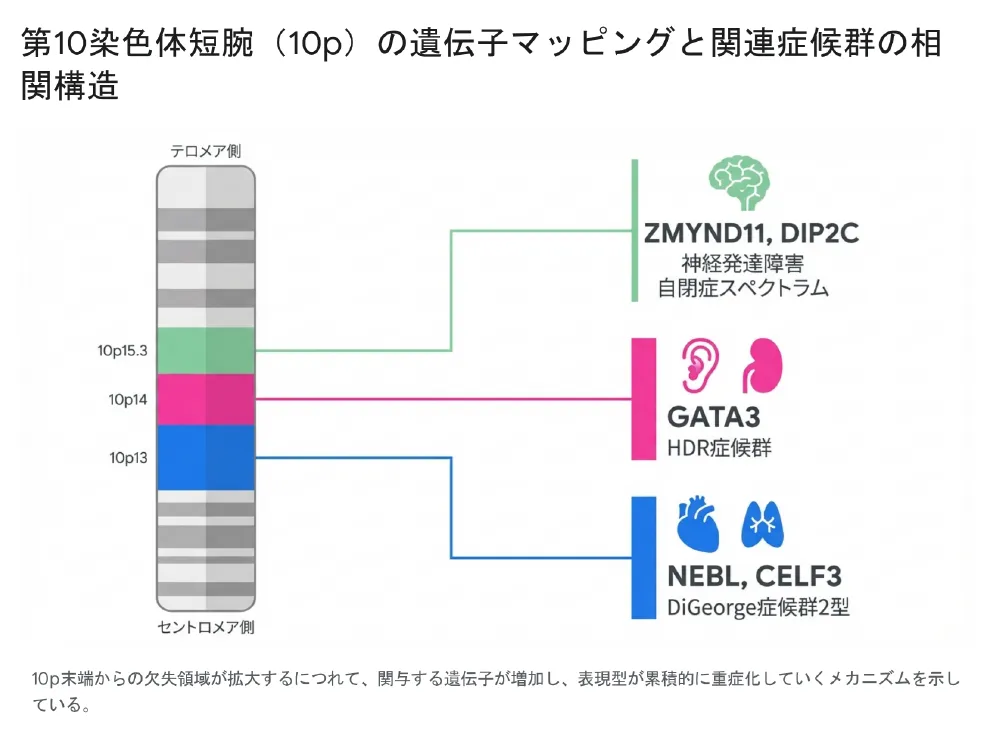
(図:10p末端からの欠失領域が拡大するにつれて、関与する遺伝子が増加し、表現型が累積的に重症化していくメカニズム)
テロメア(末端)側からセントロメア(中心)側に向かって、主要な3つの臨界領域(Critical region)とその責任遺伝子について詳しく解説します。
① 10p15.3微小欠失領域:中枢神経系発達の基盤
染色体の最末端に位置する10p15.3領域が欠失すると、主に認知機能障害、言語遅滞、自閉症スペクトラム様行動、運動発達遅滞などの中枢神経系の発達異常が引き起こされます。ここに関わる極めて重要な遺伝子がZMYND11遺伝子とDIP2C遺伝子です。
- ZMYND11遺伝子: 細胞核内でクロマチン構造を認識し、遺伝子の転写(読み取り)をコントロールする役割を持ちます。この遺伝子が機能不全に陥ると、大脳皮質における神経前駆細胞の分化異常が起こります。臨床的には「MRD30」として独立して分類されるほど、重度の知的障害や複雑な行動異常の強力な原因となります。
- DIP2C遺伝子: 脳組織において高い発現を示し、発達性ディスレクシア(読字障害)や自閉症の強力な候補遺伝子です。神経ペプチドのシグナル伝達や記憶形成経路に深く関与しています。
「常染色体優性(顕性)」とは、一対(2つ)ある遺伝子のうち、片方に変異や欠失があるだけで病気の症状が現れる遺伝形式のことです。(※かつては優性・劣性と呼んでいましたが、現在では誤解を避けるため「顕性(けんせい)」「潜性(せんせい)」という用語が推奨されています)。ZMYND11遺伝子の異常は、片方だけでも脳の発達に重大な影響を及ぼします。
② 10p14領域:HDR症候群のマスターレギュレーター
10p15領域からさらに中心へ進んだ10p14領域の欠失は、発達遅滞とは根本的に異なる、明確な多臓器形成異常症候群である「HDR症候群(Barakat症候群)」の直接的な原因となります。HDRとは、以下の3つの主要な病的徴候の頭文字をとったものです。
- Hypoparathyroidism(副甲状腺機能低下症)
- Deafness(感音難聴)
- Renal dysplasia(腎異形成)
この表現型を決定づけているのがGATA3遺伝子です。GATA3は、胎生期(お腹の中にいる時期)において、副甲状腺、内耳組織、腎臓を形作るための設計図を束ねる「現場監督(マスターレギュレーター)」として働きます。
10p14欠失によってGATA3遺伝子の片方が失われると、細胞内で作られるタンパク質の量が「正常に臓器を作るために必要な閾値(基準値)」を下回ってしまいます。このように、片方の遺伝子が失われるだけで機能不全に陥る現象を「ハプロ不全」と呼びます。現場監督が半分になることで、胎生期の臓器形成が途絶してしまいます。
③ 10p13-14領域:心臓血管構築と胸腺発生の臨界領域
欠失がさらにセントロメア側の10p13にかけて進展すると、「DiGeorge症候群2型(DGS2)」として知られる重篤な表現型が顕在化します。これは、胎生期における第3・第4咽頭嚢(将来の胸腺や副甲状腺になる組織)および心臓流出路の発生異常に起因します。
この領域の先天性心疾患の主要な原因遺伝子として有力視されているのがNEBL遺伝子です。心筋細胞の収縮機構(サルコメア)を維持する構造タンパク質をコードしており、この欠失が心筋の構造的脆弱性を招き、ファロー四徴症などの重篤な心奇形を誘発すると推測されています。また、広範なRNA転写後調節を担うCELF3遺伝子の欠失も、心臓および胸腺の発達障害に深く関与しています。
3. 臨床表現型:全身に現れる複雑な症状と病態生理
10p14-p13欠失を持つ患者さんは、前述した複数の重要遺伝子が同時に失われるため、複数の独立した疾患群が同時に発現する極めて過酷な経過をたどります。
① 内分泌系:難治性低カルシウム血症と副甲状腺形成不全
GATA3遺伝子の欠失等により、血中のカルシウム濃度を調整する「副甲状腺」が極端に低形成、あるいは完全に無形成(aplasia)となります。これにより副甲状腺ホルモン(PTH)が分泌されず、生体は慢性的な重症低カルシウム血症に陥ります。
乳幼児期には極度の不機嫌や哺乳不良として現れますが、カルシウムが急低下すると、全身の筋肉がけいれんする「テタニー」や、致命的なてんかん発作、昏睡を引き起こす非常に危険な状態となります。
② 循環器系:円錐動脈幹奇形と中隔欠損
先天性心疾患は、新生児の約半数に認められ、命に直結する最大の要因です。心室中隔欠損症(VSD)などの単純な穴だけでなく、大血管の根元に構造的異常が生じる「円錐動脈幹異常」を高い確率で合併します。具体的には、ファロー四徴症、両大血管右室起始症(DORV)、総動脈幹症などがあり、出生直後からチアノーゼや心不全を避けるための高度な外科手術が必要となります。
③ 免疫系:胸腺無形成とT細胞分化障害のスペクトラム
DiGeorge症候群2型の特徴として、免疫の学校である「胸腺」の発生異常が生じます。T細胞(外敵と戦う免疫細胞)が成熟できなくなるため、易感染性(感染症にかかりやすい状態)を示します。特に全DGS症例の約1%に見られる「完全胸腺無形成」の場合は、重症複合免疫不全症(SCID)と同様の致死的な免疫不全状態に陥ります。
⚠️ 生ワクチン接種に関する重大な警告:
T細胞機能が著しく低下している患者に対し、結核のBCGや麻疹・風疹などの「生ワクチン(弱毒化した生きたウイルス・細菌)」を接種した場合、免疫系がワクチン株を排除できず、制御不能な全身性感染症を引き起こして死亡する事例が報告されています。免疫機能の評価が終わるまで、生ワクチンの接種は絶対禁忌です。
④ 聴覚および腎・泌尿器系の異常(HDR要素)
胎生期の内耳の発達障害により、中等度から重度(両耳で80dB以上の喪失など)の感音難聴が生じます。HDR表現型を持つ患者の96.7%に認められる最も一貫した特徴であり、乳幼児期の言語獲得に重大な影響を与えます。
また、腎・泌尿器系では、腎低形成、多嚢胞腎、膀胱尿管逆流症などの構造異常が高頻度で発生し、将来的に不可逆的な慢性腎臓病(CKD)や末期腎不全へ進行するリスクを抱えます。
⑤ 中枢神経系・頭蓋顔面形態・その他の異常
10p15.3領域のZMYND11等の欠失が及ぶと、大脳皮質の萎縮、小頭症、水頭症、さらには重篤なキアリ奇形1型などの脳の構造異常が顕在化します。これにより重度の精神運動発達遅滞、難治性てんかんが発症します。
顔貌の特徴(広い鼻根部、極端に短い眼瞼裂、内眼角贅皮、下顎後退など)や、重篤な嚥下機能障害による持続的な発育不全(FTT)も高頻度で観察されます。
4. 鑑別診断:22q11.2欠失(DiGeorge症候群1型)との決定的な違い
臨床現場で「胸腺低形成、副甲状腺機能低下症、先天性心疾患」の三徴候を見た場合、最初に疑われるのは、頻度が圧倒的に高い22q11.2欠失症候群(DiGeorge症候群1型:DGS1)です。DiGeorge様症状を示す患者の90%以上は第22染色体の異常であり、第10染色体短腕の欠失(DGS2)は極めて稀なマイナーバリアントです。
DGS1の原因である「TBX1遺伝子」と、DGS2に関連する「GATA3遺伝子」は、胎生期の臓器発生において密接に連携するネットワークを形成しているため、どちらが壊れても最終的に似たような症状(フェノコピー)が引き起こされます。しかし、両者には明確な鑑別の決定打が存在します。
| 臨床的特徴・項目 | DiGeorge症候群1型 (DGS1) ※22q11.2欠失 |
DiGeorge症候群2型 (DGS2) ※10p14-p13欠失 / HDR合併 |
|---|---|---|
| 聴覚障害の特徴 | 軽度〜中等度の伝音難聴が主体(口蓋裂や反復性中耳炎による二次的障害) | GATA3欠失による内耳の原発性発生障害に起因する、重度の両側性感音難聴 |
| 腎・泌尿器系の異常 | 稀に構造異常を認めるが、通常は主症状とはならない | 重篤な腎異形成、多嚢胞腎、慢性腎臓病への進行リスクが高い(HDR要素) |
| 頭蓋顔面形態(顔貌) | 球状の鼻尖、泣き顔の非対称性、長い顔 | 広い鼻根部、極端に短い眼瞼裂、粗野な顔貌 |
| 中枢神経・発達障害 | 軽度から中等度の学習障害、精神疾患(統合失調症など)の好発 | 極めて重度な知的障害、言語の完全な欠如、脳の構造的奇形(キアリ奇形等) |
もしDiGeorge症候群が強く疑われる状況において、「重度の感音難聴」と「エコー等で確認される広範な腎異形成」が合併している場合は、単なるDGS1ではなく、10p欠失(DGS2)の可能性を極めて高く見積もる必要があります。
5. 巨大欠失(Large Deletions)がもたらす重症化リスク
10pの欠失領域が広大であるほど、表現型は単純な足し算を超えて重症化・複雑化します。過去に報告された最大の欠失例は、16Mbという広大な範囲(10p15.3から10p13にかけて)が失われた症例です。
この症例では、ZMYND11、GATA3、NEBLといった主要遺伝子に加えて、腎低形成に関連するITGA8、重症複合免疫不全症に関連するDCLRE1Cなど、多数の重篤な疾患関連遺伝子が同時に失われました。その結果、巨大なVSDを伴う両大血管右室起始症、重度の嚥下障害、難治性低カルシウム血症、重複子宮など、想定されるほぼすべての重症表現型が同時多発的に発現し、複合的なハプロ不全がいかに生体の恒常性維持機構を徹底的に破壊するかが如実に示されました。
6. 最新の遺伝子検査(CMA・NGS)の重要性
従来のFISH法(蛍光in situハイブリダイゼーション)は特定の染色体(主に22q11.2領域)のみを標的とするため、もし患者が10p欠失であった場合、検査を「すり抜けて見逃される」リスクがあります。
したがって、先天性心疾患や低カルシウム血症を呈する新生児で、標準的なFISH検査が陰性であった場合、速やかに全ゲノムを網羅的に解析可能な高解像度の染色体マイクロアレイ検査(CMA)や次世代シーケンシング(NGS)へと診断ステップを進めることが絶対的に推奨されます。
※当院での遺伝子検査について:
正確な遺伝学的診断は、その後の人生における先制的な医療介入(予防的治療)の要となります。当院では、様々な遺伝性疾患に対応する次世代シーケンシングを用いたパネル検査をご提供しています。骨格異常や多発性の奇形を伴う疾患の鑑別には、遺伝性多発性外骨腫症NGSパネルなども活用し、極めて高度な精密医療(Precision medicine)を実践しています。
7. 生涯を見据えた集学的マネジメントと治療指針
染色体の欠損そのものを治癒させる根本治療は存在しませんが、多臓器の症状に対する緻密な対症療法と、致死的な合併症を防ぐための先制的なモニタリングにより、患者さんの生命予後とQOL(生活の質)を劇的に改善することが可能です。
小児科、臨床遺伝専門医、内分泌科、心臓血管外科、腎臓内科、耳鼻咽喉科などによる高度に連携された集学的医療チームの介入が不可欠です。
低カルシウム血症の厳格なコントロールと腎機能保護
副甲状腺機能低下症による低カルシウム血症は、生後直後から最優先で内科的治療が必要です。活性型ビタミンD製剤の持続的な経口投与が中心となります。ここで極めて重要な注意点があります。
治療の落とし穴:血中カルシウム濃度を無理に正常値の真ん中まで上げようとすると、腎臓から大量のカルシウムが尿に排泄され「高尿カルシウム血症」を引き起こします。これが腎結石や腎石灰化を招き、GATA3欠失で脆弱な腎臓を完全に破壊してしまいます。治療の目標は「血中カルシウム濃度を正常範囲の下限(Low-normal range)に維持し、テタニー症状を抑えること」に留めるべきです。
先制医療としての腎臓・聴覚のモニタリング
聴覚機能の温存:診断と同時に精密なABR(聴性脳幹反応)等を実施し、難聴が確認された場合は言語発達の臨界期を逃さないよう補聴器や人工内耳の早期介入を行います。
腎機能の生涯モニタリング:腎構造異常は無症状のまま進行します。初期診断時のエコー検査はもちろん、生涯にわたり血清クレアチニン、尿蛋白、血圧の定期的なモニタリングを実施し、慢性腎臓病への進行を先制して管理します。
神経発達支援と療育的アプローチ
ZMYND11などの欠失による重度の知的・運動発達遅延には、包括的かつ早期からの療育(Early intervention program)が最大の効果を発揮します。理学療法(PT)、作業療法(OT)、言語聴覚療法(ST)を組み合わせ、患者さんが持つ潜在的な認知能力と身体機能を最大限に引き出すことを目指します。
よくある質問(FAQ)
関連記事
参考文献
- Distal deletion 10p syndrome [Orphanet]
- Deletion mapping on chromosome 10p and definition of a critical region for the second DiGeorge syndrome locus DGS2 [PubMed]
- GATA3 haplo-insufficiency causes human HDR syndrome [PubMed]
- Clinical description of a neonate carrying the largest reported 10p deletion [PMC]
- ZMYND11 functions in bimodal regulation of latent genes and brain-like splicing to safeguard corticogenesis [PMC]
- Hypoparathyroidism, Sensorineural Deafness, and Renal Disease Syndrome Presenting With Febrile Seizures and Hypocalcemia [PMC]
- Immunodeficiency in DiGeorge Syndrome and Options for Treating Cases with Complete Athymia [Frontiers]






